
La Gazette du Canada, Partie I, volume 156, numÃĐro 51 : RÃĻglement modifiant certains rÃĻglements pris en vertu de la Loi sur les aliments et drogues (homologation agile)
Le 17 dÃĐcembre 2022
Fondement lÃĐgislatif
Loi sur les aliments et drogues
MinistÃĻre responsable
MinistÃĻre de la SantÃĐ
RÃSUMà DE LâÃTUDE DâIMPACT DE LA RÃGLEMENTATION
(Le prÃĐsent rÃĐsumÃĐ ne fait pas partie du RÃĻglement.)
RÃĐsumÃĐ
Enjeux : Le rythme de lâinnovation dâaujourdâhui signifie que les drogues et les instruments mÃĐdicaux ÃĐvoluent plus rapidement que les cadres rÃĐglementaires traditionnels qui ont ÃĐtÃĐ conçus pour les rÃĐglementer. De plus, les organismes de rÃĐglementation internationaux ont ÃĐvoluÃĐ vers une plus grande surveillance aprÃĻs la mise en marchÃĐ des drogues et des instruments mÃĐdicaux. Au fil du temps, SantÃĐ Canada a adoptÃĐ des modifications lÃĐgislatives et rÃĐglementaires et a mis en Åuvre certaines pratiques au moyen de politiques visant à rÃĐgler ces enjeux. Dâautres modifications rÃĐglementaires sont nÃĐcessaires pour fournir un cadre juridique à lâappui des pratiques stratÃĐgiques et pour assurer la transparence, la prÃĐvisibilitÃĐ, lâuniformitÃĐ et la conformitÃĐ.
Bon nombre des rÃĻglements propres aux mÃĐdicaments biologiques (produits biologiques) sont trop normatifs et spÃĐcifiques aux produits et ne reflÃĻtent pas la science et la technologie actuelles. La perspective de lâabrogation des exigences propres aux produits biologiques a soulignÃĐ la nÃĐcessitÃĐ de prÃĐciser les rÃĻgles et les attentes actuelles en matiÃĻre de contrÃīle de la qualitÃĐ dans le cadre des bonnes pratiques de fabrication (BPF) qui sâappliquent à tous les produits.
Diverses sous-populations, comme les femmes, les minoritÃĐs raciales, les enfants et les personnes ÃĒgÃĐes sont souvent sous-reprÃĐsentÃĐes dans les donnÃĐes dâessais cliniques, ce qui a une incidence sur la capacitÃĐ de SantÃĐ Canada à dÃĐterminer les risques et les diffÃĐrents profils dâinnocuitÃĐ et dâefficacitÃĐ au sein de diverses sous-populations.
Enfin, les exigences actuelles ne permettent pas aux fabricants de nuancer les limites de puretÃĐ et de puissance qui sont diffÃĐrentes de celles des publications ÃĐnumÃĐrÃĐes à lâannexe B de la Loi sur les aliments et drogues (la Loi) et qui pourraient Être considÃĐrÃĐes comme acceptables par SantÃĐ Canada lorsquâune norme du fabricant est revendiquÃĐe. De plus, les fabricants de certaines drogues sont tenus dâindiquer la norme sur lâÃĐtiquette de leur drogue, ce qui est une exigence propre au Canada et a parfois crÃĐÃĐ des dÃĐfis pour les fabricants en raison de lâespace limitÃĐ disponible sur lâÃĐtiquette.
Description : SantÃĐ Canada propose de nouvelles dispositions ciblÃĐes et des modifications rÃĐglementaires au RÃĻglement sur les aliments et drogues (RAD) et au RÃĻglement sur les instruments mÃĐdicaux (RIM) qui permettraient de respecter les engagements du ministÃĻre en matiÃĻre de modernisation et de tirer parti des politiques et des pratiques de longue date. Le projet de rÃĐglementation tiendrait compte de lâexpÃĐrience rÃĐcente avec les assouplissements rÃĐglementaires mis à lâessai avec succÃĻs dans le cadre des arrÊtÃĐs dâurgence liÃĐs à la COVID-19 et de leurs transitions vers la rÃĐglementation. Le projet comprend des composantes distinctes qui :
- permettraient lâutilisation des conditions sur le numÃĐro dâidentification dâune drogue de toute drogue;
- ÃĐlargiraient la portÃĐe de lâutilisation des conditions pour les instruments mÃĐdicaux de classe II, III et IV;
- exigeraient des plans de gestion des risques (PGR) pour certaines drogues pour usage humain pour permettre de gÃĐrer les risques et les incertitudes;
- permettraient lâexamen en continu de certaines prÃĐsentations de drogues, y compris celles de drogues destinÃĐes à rÃĐpondre à une urgence de santÃĐ publique;
- clarifieraient les attentes selon lesquelles une drogue doit Être fabriquÃĐe, emballÃĐe et ÃĐtiquetÃĐe, testÃĐe et entreposÃĐe, y compris pendant le transport, dâune façon qui assure sa qualitÃĐ;
- moderniseraient les exigences relatives aux produits biologiques en abrogeant les exigences dÃĐsuÃĻtes et en les remplaçant par celles qui tiennent compte des pratiques actuelles;
- clarifieraient, dans la rÃĐglementation, le pouvoir dâexaminer certains renseignements obtenus à lâextÃĐrieur dâune prÃĐsentation de drogue nouvelle afin dâappuyer lâexamen de SantÃĐ Canada de cette prÃĐsentation de drogue nouvelle;
- exigeraient des fabricants quâils soumettent des donnÃĐes dâessais cliniques humains ventilÃĐes par des sous-groupes de population (donnÃĐes dÃĐsagrÃĐgÃĐes) pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain, telles quâelles ont ÃĐtÃĐ soumises à la Food and Drug Administration (FDA) des Ãtats-Unis ou à lâAgence europÃĐenne des mÃĐdicaments (EMA);
- mettraient à jour les exigences relatives aux normes dâÃĐtiquetage et aux exigences pour les fabricants qui revendiquent une norme dâun fabricant pour leur drogue.
Justification : Depuis plusieurs annÃĐes, SantÃĐ Canada participe activement à la modernisation lÃĐgislative et rÃĐglementaire afin dâappuyer un cadre de rÃĐglementation des drogues et des instruments mÃĐdicaux qui superviserait plus efficacement les produits rÃĐglementÃĐs tout au long de leur cycle de vie. La proposition vise à respecter les engagements en matiÃĻre de modernisation de la rÃĐglementation ÃĐnoncÃĐs dans le Secteur de la santÃĐ et des sciences biologiques : Examen rÃĐglementaire ciblÃĐ – Feuille de route rÃĐglementaire, qui appuie la rÃĐduction des irritants rÃĐglementaires et des obstacles à lâinnovation en rendant le systÃĻme de rÃĐglementation canadien fondÃĐ sur des donnÃĐes scientifiques plus agile et plus harmonisÃĐ Ã lâÃĐchelle internationale.
GrÃĒce à lâinclusion des conditions, le RÃĻglement modifiant certains rÃĻglements pris en vertu de la Loi sur les aliments et drogues (homologation agile) [le rÃĻglement proposÃĐ] permettrait au ministre de la SantÃĐ (le ministre) de mieux gÃĐrer les risques et les incertitudes liÃĐs aux drogues et aux instruments mÃĐdicaux, et de sâadapter à lâinnovation et aux changements dans les produits thÃĐrapeutiques. Lâofficialisation dans la rÃĐglementation de la pratique de longue date de SantÃĐ Canada en ce qui concerne les PGR appuierait mieux lâÃĐvaluation de lâinformation pendant la commercialisation qui pourrait avoir une incidence sur le profil avantages-risques des drogues pour usage humain. Les modifications proposÃĐes pourraient faciliter un accÃĻs anticipÃĐ au marchÃĐ pour les drogues qui sont admissibles à un examen en continu.
Les exigences en matiÃĻre de contrÃīle de la qualitÃĐ seraient clarifiÃĐes en ce qui a trait à toutes les drogues afin de complÃĐter les modifications proposÃĐes pour les produits biologiques. Les exigences propres aux produits biologiques seraient remplacÃĐes par une rÃĐglementation plus large et plus souple qui permettrait de mieux tenir compte des progrÃĻs scientifiques et technologiques et de soutenir la pratique actuelle.
Les modifications proposÃĐes clarifieraient ÃĐgalement dans la rÃĐglementation le pouvoir du ministre dâexaminer des renseignements et des documents obtenus de sources autres que la prÃĐsentation, à lâappui de lâexamen dâune nouvelle drogue, conformÃĐment à la pratique actuelle.
De plus, lâaccÃĻs à des donnÃĐes dÃĐsagrÃĐgÃĐes prÃĐsentÃĐes à la FDA des Ãtats-Unis ou à lâEMA amÃĐliorerait grandement la capacitÃĐ de SantÃĐ Canada à ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de nouvelles drogues pour usage humain dans certains sous-groupes de la population. Cette ÃĐtape vers la promotion dâune plus grande diversitÃĐ dans les essais cliniques souligne lâengagement continu de SantÃĐ Canada dâaborder les preuves croissantes des disparitÃĐs potentielles en matiÃĻre de santÃĐ pour les populations à la recherche dâÃĐquitÃĐ et les populations dÃĐtentrices de droits, comme les femmes, les minoritÃĐs raciales et ethniques ou les peuples autochtones, en raison de leur sous-reprÃĐsentation dans les donnÃĐes cliniques.
Enfin, la mise à jour du rÃĻglement sur lâÃĐtiquetage de la norme pour des drogues spÃĐcifiques et des exigences pour certaines drogues qui revendiquent une norme dâun fabricant rÃĐpondrait aux prÃĐoccupations de longue date des fabricants selon lesquelles les exigences actuelles ne sont pas toujours nÃĐcessaires pour assurer lâinnocuitÃĐ, lâefficacitÃĐ et la qualitÃĐ dâune drogue.
ÃnoncÃĐ des coÃŧts et des avantages : Les coÃŧts supplÃĐmentaires pour lâindustrie sont estimÃĐs à 158 millions de dollars (valeur actualisÃĐe [VA]). Il devrait coÃŧter à SantÃĐ Canada 26 millions de dollars (VA) pour examiner et gÃĐrer les conditions, les examens en continu et les PGR. Par consÃĐquent, le coÃŧt total prÃĐvu de la modification proposÃĐe est de 184 millions de dollars (VA) sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Ces coÃŧts seraient compensÃĐs par une autorisation de mise en marchÃĐ plus rapide, une amÃĐlioration de la qualitÃĐ de vie des Canadiens et lâÃĐlimination des exigences rÃĐglementaires relatives aux normes. Ces avantages sont estimÃĐs à 440 millions de dollars (VA) sur une pÃĐriode de 10 ans. Lâincidence nette totale prÃĐvue de cette proposition est de 256 millions de dollars (VA) en avantages nets.
RÃĻgle du ÂŦ un pour un Âŧ et lentille des petites entreprises : La modification proposÃĐe est une rÃĐduction en ce qui a trait à la rÃĻgle du ÂŦ un pour un Âŧ, car le fardeau administratif prÃĐvu pour lâindustrie devrait Être rÃĐduit de 25 177 $ (dollars de 2012) par annÃĐe, ou de 1 678 $ (dollars de 2012) par entreprise. La lentille des petites entreprises sâapplique, car il y a environ 225 petites entreprises au Canada qui pourraient Être touchÃĐes par cette proposition.
Enjeux
Le rythme de lâinnovation aujourdâhui signifie que les drogues et les instruments mÃĐdicaux ÃĐvoluent plus rapidement que les cadres rÃĐglementaires traditionnels qui ont ÃĐtÃĐ conçus pour les accommoder. Par consÃĐquent, le ministre a une capacitÃĐ limitÃĐe dâobliger les fabricants à prendre des mesures pour gÃĐrer les risques et les incertitudes associÃĐs à une drogue ou à un instrument mÃĐdical, ou pour tenir compte de diffÃĐrentes façons dans lesquelles lâinformation relative aux prÃĐsentations de drogue pourrait Être fournie ou obtenue. Bien que SantÃĐ Canada ait mis en Åuvre certaines pratiques au moyen de politiques visant à rÃĐgler ces enjeux, il est nÃĐcessaire de codifier de telles pratiques et de promouvoir la conformitÃĐ au moyen de mesures qui assurent la transparence, la prÃĐvisibilitÃĐ et lâuniformitÃĐ.
En ce qui concerne la fabrication, lâemballage et lâÃĐtiquetage, la vÃĐrification, lâentreposage et le transport dâune drogue, malgrÃĐ le fait que le RAD attribue aux services de contrÃīle de la qualitÃĐ des responsabilitÃĐs en ce qui concerne les procÃĐdures et les mÃĐthodes, il nây a pas dâexigence claire que de telles activitÃĐs doivent Être menÃĐes de maniÃĻre à maintenir et à assurer la qualitÃĐ de la drogue. Un exemple des lacunes existantes est lâattente que les drogues doivent Être entreposÃĐes selon leurs conditions dâentreposage approuvÃĐes.
De plus, bon nombre des dispositions relatives aux produits biologiques ne reflÃĻtent pas la science actuelle, la technologie et les façons dont lâindustrie a ÃĐvoluÃĐ au cours des 70 derniÃĻres annÃĐes. Bon nombre des dispositions sont trop prescriptives et spÃĐcifiques aux produits et ne sont plus scientifiquement pertinentes. Cela constitue un risque pour la santÃĐ et la sÃĐcuritÃĐ des Canadiens que SantÃĐ Canada aborde actuellement au moyen de pratiques et de politiques souples et axÃĐes sur les rÃĐsultats.
Lorsquâil examine une prÃĐsentation de drogue, afin que le ministre puisse prendre la dÃĐcision la plus ÃĐclairÃĐe au sujet de lâautorisation de la drogue, le ministre peut tenir compte des renseignements qui pourraient provenir dâautres sources que la prÃĐsentation. à lâheure actuelle, les sources dâinformation explicitement autorisÃĐes par la rÃĐglementation sont trop limitÃĐes et ne comprennent pas les sources clÃĐs de matÃĐriel et dâinformation qui pourraient Être nÃĐcessaires pour que SantÃĐ Canada ÃĐvalue et autorise une nouvelle drogue.
De plus, SantÃĐ Canada nâa pas actuellement besoin de recevoir des donnÃĐes dÃĐsagrÃĐgÃĐes pour appuyer lâÃĐvaluation des prÃĐsentations de drogues nouvelles ou de supplÃĐments à une prÃĐsentation de drogue nouvelle. Cependant, la recherche et les donnÃĐes probantes suggÃĻrent quâil peut y avoir diffÃĐrents profils dâefficacitÃĐ et dâinnocuitÃĐ pour certaines drogues au sein de diverses sous-populations, y compris celles qui sont frÃĐquemment sous-reprÃĐsentÃĐes dans les essais cliniques (par exemple les femmes, les minoritÃĐs raciales, les enfants et les personnes ÃĒgÃĐes). La rÃĐception de donnÃĐes dÃĐsagrÃĐgÃĐes permettrait à SantÃĐ Canada de mieux ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ dâune drogue au sein de diverses sous-populations et de dÃĐterminer si elle peut prÃĐsenter un risque accru pour une certaine sous-population comparativement à dâautres.
Enfin, dans les cas oÃđ une norme dâun fabricant est revendiquÃĐe, le RAD ne permet pas aux fabricants de drogues de fixer des limites de puretÃĐ et/ou de puissance qui sont diffÃĐrentes de celles des publications ÃĐnumÃĐrÃĐes à lâannexe B de la Loi sur les aliments et drogues (la Loi) avec des donnÃĐes supplÃĐmentaires qui pourraient Être considÃĐrÃĐes comme acceptables par SantÃĐ Canada. Les fabricants ont indiquÃĐ que cette exigence a occasionnellement donnÃĐ lieu au retrait dâune drogue du marchÃĐ au Canada. De plus, les exigences en matiÃĻre dâÃĐtiquetage relatives à la norme dâune drogue sont propres au Canada (câest-à -dire quâelles ne sont pas exigÃĐes par dâautres organismes de rÃĐglementation, comme la FDA et lâEMA) et ont parfois crÃĐÃĐ des dÃĐfis pour certains fabricants en raison de lâespace limitÃĐ disponible sur lâÃĐtiquette.
Contexte
Le ministre de la SantÃĐ est responsable des activitÃĐs rÃĐglementaires liÃĐes à lâinnocuitÃĐ, à lâefficacitÃĐ et à la qualitÃĐ des drogues et des instruments mÃĐdicaux. Le pouvoir du ministre dÃĐcoule de la Loi et des rÃĻglements pris en application de celle-ci, y compris le RAD et le RIM.
Depuis plusieurs annÃĐes, SantÃĐ Canada participe activement à la modernisation lÃĐgislative et rÃĐglementaire afin dâappuyer des cadres de rÃĐglementation des drogues et des instruments mÃĐdicaux qui supervisent plus efficacement les produits rÃĐglementÃĐs tout au long de leur cycle de vie. En 2014, le Parlement a adoptÃĐ la Loi de Vanessa, qui a modifiÃĐ la Loi pour inclure les pouvoirs de rÃĐglementation aux fins de la collecte de renseignements sur lâinnocuitÃĐ concernant les drogues et les instruments mÃĐdicaux, aussi appelÃĐs produits thÃĐrapeutiques, et les pouvoirs qui permettent au ministre de prendre des mesures en cas dâidentification dâun risque grave pour la santÃĐ. Il sâagissait dâune ÃĐtape clÃĐ de lâinitiative de SantÃĐ Canada visant à moderniser la Loi et ses rÃĻglements connexes rÃĐgissant les produits thÃĐrapeutiques. Les rÃĻglements pris en vertu des nouveaux pouvoirs de la Loi de Vanessa ont introduit des mesures qui ont :
- renforcÃĐ la surveillance de lâinnocuitÃĐ des produits thÃĐrapeutiques tout au long de leur cycle de vie (par exemple les conditions pour les opioÃŊdes et les drogues dÃĐsignÃĐes contre la COVID-19);
- amÃĐliorÃĐ la surveillance aprÃĻs la mise en marchÃĐ (par exemple les dÃĐclarations obligatoires des rÃĐactions graves aux mÃĐdicaments et des incidents liÃĐs aux instruments mÃĐdicaux par les hÃīpitaux, les dÃĐclarations des mesures rÃĐglementaires ÃĐtrangÃĻres comportant un risque grave pour la santÃĐ humaine);
- ÃĐlargi les autoritÃĐs post-commercialisation pour les instruments mÃĐdicaux;
- favorisÃĐ une plus grande confiance dans la surveillance des produits thÃĐrapeutique en augmentant la transparence (par exemple la diffusion publique de renseignements sur les essais cliniques);
- harmonisÃĐ les exigences avec celles des organismes de rÃĐglementation dâautres administrations.
Dâautres travaux sur la dÃĐtermination et la rÃĐponse aux irritants rÃĐglementaires et aux obstacles à lâinnovation ont ÃĐtÃĐ menÃĐs dans le cadre de lâexamen sectoriel de la rÃĐglementation du secteur de la santÃĐ et des sciences biologiques de 2019 et ont abouti au lancement du programme dâinnovation de la rÃĐglementation de SantÃĐ Canada. GrÃĒce à ces initiatives, SantÃĐ Canada sâest engagÃĐ Ã moderniser les rÃĻglements applicables afin dâoffrir au bout du compte des cadres rÃĐglementaires modernes et souples pour les drogues et les instruments mÃĐdicaux.
Certaines de ces souplesses (conditions et examens en continu) ont ÃĐtÃĐ mises à lâessai dans le cadre de la rÃĐponse à la pandÃĐmie de COVID-19 au moyen des projets rÃĐglementaires suivants :
- lâArrÊtÃĐ dâurgence concernant lâimportation, la vente et la publicitÃĐ de drogues à utiliser relativement à la COVID-19 (septembre 2020), suivi des modifications au RAD (mars 2021);
- trois arrÊtÃĐs dâurgence concernant lâimportation et la vente dâinstruments mÃĐdicaux à utiliser en regard à la COVID-19 (mars 2020, mars 2021, fÃĐvrier 2022).
Cette initiative rÃĐglementaire fait partie du programme dâinnovation de la rÃĐglementation de SantÃĐ Canada et contribuera directement à la StratÃĐgie en matiÃĻre de biofabrication et de sciences de la vie du gouvernement qui reconnaÃŪt lâimportance du systÃĻme de rÃĐglementation en tant que catalyseur de la croissance du secteur national de la biofabrication.
Conditions
Le rythme rapide de lâinnovation de lâindustrie a entraÃŪnÃĐ des incertitudes et des risques en ce qui concerne les produits thÃĐrapeutiques qui pourraient ne pas Être gÃĐrÃĐs adÃĐquatement au moyen des dispositions rÃĐglementaires existantes. Une condition est une obligation que le ministre peut imposer au titulaire dâune autorisation de produit thÃĐrapeutique de mener une activitÃĐ Ã lâÃĐgard de la drogue ou de lâinstrument mÃĐdical visÃĐ par lâautorisation. Lâobjectif principal des conditions est dâoptimiser les avantages et de gÃĐrer les risques et les incertitudes associÃĐs à la drogue ou à lâinstrument mÃĐdical, y compris en recueillant des renseignements supplÃĐmentaires aprÃĻs son autorisation. SantÃĐ Canada reconnaÃŪt depuis longtemps la valeur dâune telle approche. En 1998, la politique sur les Avis de conformitÃĐ avec conditions (AC-C) a ÃĐtÃĐ mise en place pour permettre aux fabricants de certaines drogues pour usage humain — à savoir, celles destinÃĐes à traiter une maladie ou une affection grave qui met en danger la vie ou est sÃĐvÃĻrement dÃĐbilitante — de demander une autorisation de vendre une drogue en fonction des preuves cliniques prometteuses qui indiquent que son utilisation est raisonnablement susceptible de produire le rÃĐsultat voulu. Cette politique a permis à SantÃĐ Canada de gÃĐrer plus efficacement les incertitudes liÃĐes aux risques et aux avantages et, par consÃĐquent, de faciliter une mise sur le marchÃĐ plus rapide des drogues pouvant sauver des vies lorsque le fabricant accepte dâentreprendre des ÃĐtudes plus poussÃĐes pour confirmer lâefficacitÃĐ et surveiller lâinnocuitÃĐ de ces drogues et fournir cette information au ministre aprÃĻs son entrÃĐe sur le marchÃĐ canadien.
à lâheure actuelle, le ministre a un vaste pouvoir dâimposer ou de modifier les conditions relatives aux opioÃŊdes et aux drogues dÃĐsignÃĐes contre la COVID-19 dans le RAD. Depuis lâentrÃĐe en vigueur du RIM en 1998, le ministre a obtenu le pouvoir dâimposer ou de modifier les conditions relatives à la vÃĐrification sur les licences dâinstruments mÃĐdicaux de classe II, III et IV. De plus, de grands pouvoirs en matiÃĻre de conditions ont ÃĐtÃĐ inclus dans les arrÊtÃĐs dâurgence liÃĐs à lâautorisation dâinstruments mÃĐdicaux destinÃĐs à Être utilisÃĐs à lâÃĐgard de la COVID-19.
Plans de gestion des risques
Un PGR rÃĐsume les risques et les incertitudes cernÃĐs et potentiels liÃĐs à une drogue et aux activitÃĐs de pharmacovigilance connexes, ainsi que les autres mesures que le fabricant a lâintention de mettre en place pour gÃĐrer ces risques et incertitudes.
à lâÃĐchelle internationale, la plupart des organismes de rÃĐglementation des drogues pour usage humain, y compris ceux de lâUnion europÃĐenne, du Royaume-Uni et des Ãtats-Unis, ont introduit des exigences prÃĐvues par la loi pour les PGR ou leur ÃĐquivalent. Depuis 2009, afin de mieux sâharmoniser avec la pratique internationale, SantÃĐ Canada a demandÃĐ aux fabricants de prÃĐsenter volontairement des PGR au moment du dÃĐpÃīt dâune prÃĐsentation de drogue nouvelle pour certaines nouvelles substances activesrÃĐfÃĐrence 1. En 2015, SantÃĐ Canada a publiÃĐ la Ligne directrice – PrÃĐsentation des plans de gestion des risques et des engagements en matiÃĻre de suivi qui formaliseraient la pratique de demander des PGR aux fabricants de :
- produits pharmaceutiques (y compris ceux vendus sur ordonnance et ceux en vente libre) qui comprennent de nouvelles substances actives;
- produits biologiques (y compris les produits biotechnologiques, les vaccins ainsi que les produits sanguins obtenus par fractionnement) conformÃĐment à lâannexe D de la Loi;
- produits pharmaceutiques radioactifs tels quâils sont ÃĐnoncÃĐs à lâannexe C de la Loi.
La ligne directrice indique que les fabricants devraient inclure des sections propres au Canada qui aborderaient les facteurs applicables à la population canadienne, selon le contexte et le marchÃĐ canadiens.
Examens en continu
Un ÂŦ examen en continu Âŧ permet à un fabricant de soumettre sa prÃĐsentation de drogue avec certains, mais pas tous les renseignements nÃĐcessaires pour que lâorganisme de rÃĐglementation puisse ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ dâune drogue. Pourvu que certaines conditions soient respectÃĐes, une telle prÃĐsentation peut Être dÃĐposÃĐe, ÃĐtant entendu que les renseignements manquants devraient Être fournis dans un dÃĐlai raisonnable. Toutefois, peu importe la quantitÃĐ de renseignements qui est incluse au moment du dÃĐpÃīt de la prÃĐsentation, la dÃĐcision dâautoriser la drogue ne peut Être prise que lorsque tous les renseignements requis, y compris les renseignements manquants, ont ÃĐtÃĐ fournis et jugÃĐs acceptables.
SantÃĐ Canada a utilisÃĐ des examens en continu pour accroÃŪtre lâaccÃĻs en temps opportun aux mÃĐdicaments nÃĐcessaires. Actuellement, des examens en continu sont disponibles par pratique pour les mises à jour annuelles des vaccins contre la grippe saisonniÃĻre ainsi que pour lâexamen simultanÃĐ des prÃĐsentations de drogue pour usage vÃĐtÃĐrinaire avec les Ãtats-Unis dans le cadre du Conseil de coopÃĐration Canada–Ãtats-Unis en matiÃĻre de rÃĐglementation. Des examens en continu sont ÃĐgalement disponibles dans le RAD pour les prÃĐsentations de drogue nouvelle pour des drogues dÃĐsignÃĐes contre la COVID-19 tel quâil est modifiÃĐ par le RÃĻglement modifiant le RÃĻglement sur les aliments et drogues (ArrÊtÃĐ dâurgence concernant lâimportation, la vente et la publicitÃĐ de drogues à utiliser relativement à la COVID-19).
Assurer la qualitÃĐ des mÃĐdicaments pendant la fabrication
Les BPF sont un systÃĻme dâassurance de la qualitÃĐ reconnu à lâÃĐchelle internationale qui permet de sâassurer que les drogues sont fabriquÃĐes, emballÃĐes, ÃĐtiquetÃĐes, analysÃĐes, importÃĐes, distribuÃĐes et vendues en gros de façon uniforme. Toutefois, bien quâil soit prÃĐvu que les drogues soient fabriquÃĐes, emballÃĐes, ÃĐtiquetÃĐes, analysÃĐes et entreposÃĐes, y compris pendant le transport, dâune maniÃĻre qui assurerait la qualitÃĐ de lâingrÃĐdient actif et de la drogue sous forme posologique, cela nâest pas clairement ÃĐnoncÃĐ dans les textes rÃĐglementaires.
Modernisation des exigences relatives aux produits biologiques
Les produits biologiques sont fabriquÃĐs et extraits de tissus ou dâorganismes vivants, certains avec des gÃĻnes modifiÃĐs. Leur innocuitÃĐ et leur efficacitÃĐ dÃĐpendent grandement de leurs matÃĐriaux sources (par exemple les cellules, les organismes vivants) et des matÃĐriaux auxiliaires (par exemple les additifs utilisÃĐs pour complÃĐter le milieu de culture cellulaire). De plus, les produits biologiques ont tendance à Être des solutions stÃĐriles et injectables qui traitent des affections graves ou mettant en danger la vie, et les risques liÃĐs à ces drogues peuvent Être tout aussi graves ou mettre en danger la vie.
Une grande partie du titre 4 de la partie C du RAD consiste en des rÃĻglements propres aux produits qui ont ÃĐtÃĐ adoptÃĐs dans les annÃĐes 1950 et 1960 pour rÃĐpondre aux questions de lâÃĐpoque. Ãtant donnÃĐ que le nombre de produits biologiques a considÃĐrablement augmentÃĐ au fil du temps et que les rÃĻglements ont toujours ÃĐtÃĐ essentiellement axÃĐs sur les produits, ils nâont pas suivi le rythme des progrÃĻs scientifiques.
Des modifications importantes ont ÃĐtÃĐ apportÃĐes en 1997 afin de fusionner les exigences relatives aux licences dâÃĐtablissement pour la fabrication de mÃĐdicaments pharmaceutiques, de produits biologiques et de produits pharmaceutiques radioactifs dans le titre 1A du RAD. Bien que ces modifications aient introduit des inspections de BPF liÃĐes aux locaux, au personnel, aux processus et aux produits, SantÃĐ Canada a maintenu, en vertu de la politique, la capacitÃĐ dâutiliser les renseignements obtenus sur place, afin de confirmer la capacitÃĐ du fabricant à produire rÃĐguliÃĻrement un produit biologique sÃŧr et à vÃĐrifier les renseignements fournis dans la prÃĐsentation connexe.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue
ConformÃĐment à la pratique susmentionnÃĐe, SantÃĐ Canada utilise lâinformation provenant de diverses sources, y compris les ÃĐvaluations sur place (ESP) et les inspections de BPF, dans lâexamen dâune prÃĐsentation de drogue, afin dâÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de la drogue. SantÃĐ Canada examine actuellement les renseignements et les documents disponibles, y compris ceux obtenus à lâextÃĐrieur de la prÃĐsentation, afin de prendre une dÃĐcision ÃĐclairÃĐe quant à lâautorisation dâune drogue pour le marchÃĐ canadien.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain
La collecte de donnÃĐes dÃĐsagrÃĐgÃĐes sur les participants aux essais cliniques est une mesure clÃĐ que SantÃĐ Canada peut prendre pour dâabord ÃĐvaluer et, au besoin, traiter tout risque et avantage divergents des drogues dans diverses sous-populations. SantÃĐ Canada accorde de lâimportance à lâÃĐlaboration et à la conduite dâessais cliniques inclusifs, particuliÃĻrement pour les populations sous-reprÃĐsentÃĐes. Les demandes prÃĐsentÃĐes à SantÃĐ Canada comportent gÃĐnÃĐralement un certain degrÃĐ de donnÃĐes dÃĐmographiques dÃĐsagrÃĐgÃĐes; toutefois, elles ne reflÃĻtent pas toujours le nombre de patients ou ne sont pas ventilÃĐes par sous-groupes de population (par exemple ÃĒge, sexe, genre ou race).
Normes
Les normes sur les drogues comprennent des critÃĻres et des mÃĐthodes dâessai qui aident à assurer la qualitÃĐ dâune drogue. Ces normes peuvent Être prescrites par rÃĻglement, dÃĐfinies dans une pharmacopÃĐerÃĐfÃĐrence 2 inscrite à lâannexe B de la Loi ou ÃĐlaborÃĐes par le fabricant de la drogue. Si une norme nâa pas ÃĐtÃĐ prescrite par rÃĻglement, mais quâelle figure dans une pharmacopÃĐe inscrite à lâannexe B de la Loi, un fabricant peut utiliser la norme de la pharmacopÃĐe ou sa propre norme de fabricant. Si la norme du fabricant est utilisÃĐe, le RAD exige que la drogue respecte le degrÃĐ maximal de puretÃĐ et la variation de puissance la plus petite pour cette drogue de toutes les pharmacopÃĐes inscrites à lâannexe B. Cela sâapplique à toutes les drogues, y compris les ingrÃĐdients actifs, rÃĐglementÃĐs en vertu des titres 1, 3, 4 ou 8 de la partie C du RAD tout au long de leur cycle de vie. Cette exigence prescriptive ne permet pas à ceux qui prÃĐtendent une norme du fabricant de justifier scientifiquement des contrÃīles plus ÃĐtendus de puretÃĐ et de puissance qui pourraient ÃĐgalement Être acceptables pour le ministre.
Pour les drogues rÃĐglementÃĐes en vertu des titres 1 ou 8 du RAD, le RÃĻglement exige actuellement que les ÃĐtiquettes intÃĐrieure et extÃĐrieure indiquent si le fabricant a utilisÃĐ une norme prescrite dans le RAD, une norme de lâannexe B (par exemple la U.S. Pharmacopeia) ou une norme du fabricant. Lâinclusion de cette information est une exigence propre au Canada et a parfois crÃĐÃĐ des dÃĐfis pour les fabricants en raison de lâespace limitÃĐ disponible sur lâÃĐtiquette.
Objectif
Lâobjectif des modifications rÃĐglementaires proposÃĐes est dâaccroÃŪtre la souplesse rÃĐglementaire pour suivre le rythme de lâinnovation et faciliter lâaccÃĻs à des traitements de pointe et à des thÃĐrapies prometteuses, tout en continuant de sâassurer que les drogues autorisÃĐes et les instruments mÃĐdicaux homologuÃĐs sont sÃŧrs, efficaces et assujettis à une surveillance appropriÃĐe.
Description
Ces modifications rÃĐglementaires proposÃĐes introduiraient une sÃĐrie de changements ciblÃĐs au RAD et au RIM qui reflÃĻtent les politiques existantes ainsi que les expÃĐriences rÃĐcentes de SantÃĐ Canada avec les arrÊtÃĐs dâurgence relatifs à la COVID-19.
Conditions — drogues et instruments mÃĐdicaux (modifications au RAD et au RIM)
Les modifications proposÃĐes permettraient au ministre dâimposer des conditions sur lâidentification numÃĐrique de drogue de toute drogue ou sur la licence dâun instrument mÃĐdical de classe II, III ou IV au moment de la dÃĐlivrance de lâhomologation ou à tout moment ultÃĐrieur. Les conditions pourraient ÃĐgalement Être modifiÃĐes ou supprimÃĐes en tout temps, au besoin. Bien que le RIM permette actuellement au ministre dâimposer des conditions relatives aux essais sur les licences dâinstruments mÃĐdicaux de classe II, III ou IV, les modifications proposÃĐes ÃĐlargiraient la portÃĐe des conditions qui peuvent Être imposÃĐes et leur permettraient dâÊtre exÃĐcutoires en vertu de lâarticle 21.7 de la Loi.
Avant dâimposer ou de modifier les conditions, le ministre dÃĐterminerait si les conditions contribuent à lâatteinte des objectifs de gestion des incertitudes liÃĐes aux avantages et aux risques, dâoptimisation des avantages et de gestion des risques, et de dÃĐtermination des changements liÃĐs aux avantages et aux risques de la drogue ou de lâinstrument mÃĐdical.
De plus, le ministre se demanderait si les conditions sont techniquement rÃĐalisables, sâil existe des moyens moins lourds dâatteindre ces objectifs et si elles pourraient Être atteintes par lâapplication des exigences existantes en vertu de la Loi ou du RAD et du RIM, selon le cas.
Toutes les prÃĐsentations de drogues doivent avoir les renseignements et les donnÃĐes nÃĐcessaires pour ÃĐtablir lâinnocuitÃĐ, lâefficacitÃĐ et la qualitÃĐ dâune drogue afin de dÃĐmontrer un profil avantages/risques favorable et de satisfaire à toutes les exigences en vertu du RAD pour lâautorisation de mise en marchÃĐ. Lâobjectif gÃĐnÃĐral de lâimposition des conditions serait de veiller à ce quâune drogue maintienne un profil avantages/risques favorable tout au long de son cycle de vie et à ce que lâinnocuitÃĐ, lâefficacitÃĐ ou la qualitÃĐ de la drogue nâait pas changÃĐ par rapport au moment oÃđ lâautorisation de mise sur le marchÃĐ a ÃĐtÃĐ dÃĐlivrÃĐe.
Les conditions pourraient, par exemple, Être appliquÃĐes afin de :
- gÃĐrer les incertitudes liÃĐes aux avantages et/ou aux risques cernÃĐs au moment de leur dÃĐlivrance (par exemple demander des ÃĐtudes pour confirmer les avantages dâune drogue ou dâun instrument mÃĐdical, y compris dans les sous-populations, au besoin);
- gÃĐrer les risques ou les incertitudes ÃĐmergents qui ont fait surface aprÃĻs la mise en marchÃĐ et qui nâont peut-Être pas ÃĐtÃĐ cernÃĐs avant lâautorisation;
- recueillir des donnÃĐes pour Être en mesure dâidentifier et dâÃĐvaluer les changements potentiels à lâinnocuitÃĐ et/ou à lâefficacitÃĐ et de gÃĐrer les incertitudes (par exemple pour les nouvelles technologies ou les nouvelles indications dâutilisation);
- exiger que le titulaire dâune licence dâinstrument mÃĐdical apporte des changements à lâinstrument mÃĐdical afin de continuer à maintenir lâinnocuitÃĐ et lâefficacitÃĐ.
Initialement, dans le cas des drogues, les modifications proposÃĐes seraient limitÃĐes à toute drogue dâurgence de santÃĐ publique, câest-à -dire une drogue contre la COVID-19 ou une condition dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au Canada, qui serait incorporÃĐe par rÃĐfÃĐrence. Toutefois, un an aprÃĻs lâenregistrement du projet de rÃĻglement, ces dispositions limitÃĐes seraient abrogÃĐes et les modifications proposÃĐes concernant les conditions pour toutes les drogues entreraient en vigueur. Les modifications proposÃĐes concernant les conditions pour les instruments mÃĐdicaux de classe II, III et IV entreraient ÃĐgalement en vigueur à ce moment-là .
Plans de gestion des risques (modifications au RAD)
Les modifications proposÃĐes reflÃĐteraient la pratique de longue date de SantÃĐ Canada ÃĐnoncÃĐe dans la ligne directrice intitulÃĐe PrÃĐsentation des plans de gestion des risques et des engagements en matiÃĻre de suivi. Le rÃĻglement exigerait quâun PGR soit dÃĐposÃĐ avec une demande dâidentification numÃĐrique de drogue, une prÃĐsentation de drogue nouvelle ou dâune prÃĐsentation de drogue nouvelle abrÃĐgÃĐe pour une drogue pour usage humain si le ministre a des motifs raisonnables de croire quâil y a un degrÃĐ dâincertitude important à lâÃĐgard des risques associÃĐs à la drogue, ou si la drogue prÃĐsente un risque grave de prÃĐjudice à la santÃĐ humaine qui justifie la prise de mesures au-delà des changements apportÃĐs à lâÃĐtiquette, afin de rÃĐduire la possibilitÃĐ ou la gravitÃĐ dâun tel prÃĐjudice. Les PGR seraient requis pour toutes les prÃĐsentations de drogues nouvelles pour usage exceptionnel.
Les modifications confÃĐreraient ÃĐgalement au ministre le pouvoir rÃĐglementaire dâexiger un PGR pour une drogue pour usage humain autorisÃĐe à lâÃĐgard de laquelle un PGR nâa pas encore ÃĐtÃĐ fourni lorsque les circonstances dÃĐcrites ci-dessus se prÃĐsentent.
De plus, le titulaire de lâautorisation serait tenu de fournir au ministre un PGR mis à jour si le PGR mis à jour est considÃĐrablement diffÃĐrent du PGR soumis antÃĐrieurement, ou si le ministre a des motifs raisonnables de croire quâun PGR mis à jour est requis pour rÃĐpondre à des diffÃĐrences importantes dans :
- les risques et les incertitudes associÃĐs à la drogue;
- les mesures justifiÃĐes pour gÃĐrer un risque grave de prÃĐjudice à la santÃĐ humaine.
Pour appuyer la transparence rÃĐglementaire, les modifications crÃĐeraient ÃĐgalement une nouvelle obligation pour les fabricants de soumettre un rÃĐsumÃĐ des PGR nouveaux et mis à jour dans les deux langues officielles. En pratique, ces rÃĐsumÃĐs seraient publiÃĐs en ligne et leur contenu serait harmonisÃĐ avec celui dâautres administrations avec lâajout de contenu canadien requis, le cas ÃĐchÃĐant.
Les modifications proposÃĐes relatives aux PGR ne sâappliqueraient pas aux mÃĐdicaments vÃĐtÃĐrinaires.
Examens en continu (modifications au RAD)
Les modifications proposÃĐes introduiraient un certain nombre dâoptions dâexamen en continu dans le rÃĻglement pour les prÃĐsentations de drogues nouvelles et les supplÃĐments à une prÃĐsentation de drogue nouvelle dans les cas suivants :
- la nouvelle drogue rÃĐpond à certaines conditions dÃĐcrites ci-dessous;
- la drogue est un mÃĐdicament vÃĐtÃĐrinaire et le ministre a indiquÃĐ son intention de mener lâexamen avec un organisme de rÃĐglementation ÃĐtranger;
- la souche à laquelle se rapporte un vaccin contre la grippe, figurant sur une liste incorporÃĐe par renvoi, a changÃĐ;
- la drogue est liÃĐe à une condition qui menace la santÃĐ publique figurant sur une liste incorporÃĐe par renvoi.
Ces options auraient pour effet de fournir aux fabricants qui demandent un avis de conformitÃĐ (AC) des exigences modifiÃĐes relatives à la prÃĐsentation de renseignements au moment du dÃĐpÃīt. Toute information manquante nÃĐcessaire pour ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de la drogue doit Être fournie dans un dÃĐlai raisonnable afin de permettre à SantÃĐ Canada de prendre une dÃĐcision au sujet de lâÃĐmission dâun AC. Toutes les autres exigences du RAD, comme la conservation des dossiers et la protection des donnÃĐes, ainsi que les rÃĐgimes de propriÃĐtÃĐ intellectuelle en vertu de la Loi sur les brevets, sâappliqueraient ÃĐgalement aux prÃĐsentations et aux drogues qui font lâobjet dâun examen en continu.
Drogues qui rÃĐpondent à certaines conditions
Le rÃĻglement proposÃĐ crÃĐerait un nouveau processus facultatif pour les prÃĐsentations de drogue nouvelle et pour les supplÃĐments aux prÃĐsentations de drogue nouvelle afin de faciliter lâaccÃĻs en temps opportun à de nouvelles drogues qui rÃĐpondent à une ou plusieurs des conditions suivantes :
- les mÃĐdicaments pour usage humain et vÃĐtÃĐrinaire nÃĐcessaires pour lutter contre les maladies infectieuses ÃĐmergentes qui posent ou peuvent poser un risque grave de prÃĐjudice à la santÃĐ;
- les mÃĐdicaments pour usage humain et vÃĐtÃĐrinaire destinÃĐs au traitement, Ã la prÃĐvention, Ã lâattÃĐnuation ou au diagnostic de maladies ou dâaffections graves lorsque :
- lâobjectif et les conditions dâutilisation recommandÃĐs de la nouvelle drogue ne sont pas conformes à lâobjectif et aux conditions dâutilisation recommandÃĐs de toute autre drogue pour laquelle une identification numÃĐrique de drogue a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe,
- la nouvelle drogue est beaucoup plus efficace, ou possÃĻde un risque beaucoup plus faible, comparativement à toutes les autres drogues pour les mÊmes fins et les mÊmes conditions dâutilisation recommandÃĐes pour lesquelles une identification numÃĐrique de drogue a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe.
Une ÃĐvaluation prÃĐalable à la prÃĐsentation visant à dÃĐterminer lâadmissibilitÃĐ dâun examen en continu lorsque la nouvelle drogue satisfait à une ou plusieurs des conditions ci-dessus exigerait que le fabricant :
- fournisse à SantÃĐ Canada des renseignements qui dÃĐmontrent que la drogue proposÃĐe satisfait à une ou plusieurs des conditions dÃĐcrites ci-dessus et que le fabricant possÃĻde une quantitÃĐ importante de preuves pour ÃĐtablir lâinnocuitÃĐ et lâefficacitÃĐ clinique de la drogue aux fins et aux conditions dâutilisation recommandÃĐes;
- fournisse un plan qui prÃĐcise comment et quand, dans les 155 jours suivant le dÃĐpÃīt de la prÃĐsentation, il soumettra toute information manquante. Le plan devrait envisager de fournir les renseignements manquants dans quelques transactions.
Sâil est dÃĐterminÃĐ que la prÃĐsentation de drogue proposÃĐe, pour laquelle une ÃĐvaluation prÃĐalable à la prÃĐsentation a ÃĐtÃĐ effectuÃĐe, est admissible à un examen en continu, le fabricant serait avisÃĐ par le ministre. Lâavis indiquerait une date limite de 60 jours suivant la date à laquelle lâavis est ÃĐmis par le ministre pour fournir la prÃĐsentation, ainsi que la façon et le moment oÃđ les renseignements manquants doivent Être fournis.
Afin de permettre à SantÃĐ Canada de commencer lâexamen de ces prÃĐsentations de drogues, les modifications proposÃĐes ÃĐtabliraient ÃĐgalement des exigences minimales en matiÃĻre dâinformation qui doivent Être respectÃĐes au moment du dÃĐpÃīt de la prÃĐsentation, y compris que la prÃĐsentation doit, au moment du dÃĐpÃīt, inclure une quantitÃĐ importante de renseignements pour permettre au ministre de commencer à ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de la drogue. Au moment du dÃĐpÃīt, une prÃĐsentation de drogue faisant lâobjet dâun examen en continu doit ÃĐgalement comprendre tous les renseignements normalement requis concernant la formulation, lâingrÃĐdient mÃĐdicinal, lâutilisation et la forme posologique de la drogue.
En ce qui concerne une prÃĐsentation de drogue nouvelle, les renseignements qui pourraient Être fournis à la suite de la transaction initiale comprendraient certains renseignements manquants concernant les tests et les preuves permettant dâÃĐtablir lâinnocuitÃĐ et lâefficacitÃĐ de la nouvelle drogue, des renseignements concernant les chercheurs auxquels la nouvelle drogue a ÃĐtÃĐ vendue, des preuves que les lots dâessai de la nouvelle drogue ont ÃĐtÃĐ fabriquÃĐs dâune façon qui est reprÃĐsentative de la production commerciale, le dÃĐlai dâattente de la nouvelle drogue chez les animaux destinÃĐs à lâalimentation, un plan de gestion des risques et tout rÃĐsumÃĐ et rapport de section pour toute ÃĐtude qui nâest pas encore achevÃĐe. En ce qui concerne un supplÃĐment à une prÃĐsentation de drogue nouvelle, qui se limite à des questions sensiblement diffÃĐrentes par rapport à la prÃĐsentation originale, lâoption dâun examen en continu exigerait quâune quantitÃĐ importante des renseignements soit fournie dans la transaction initiale. Cependant, cette option permettrait la soumission subsÃĐquente de toute information manquante dont le ministre a besoin pour ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de la nouvelle drogue, ainsi que tout rÃĐsumÃĐ et rapport de section pour toute ÃĐtude qui nâest pas encore achevÃĐe.
Tous les renseignements normalement requis relativement à une prÃĐsentation de drogue devraient Être fournis avant que le ministre puisse terminer lâexamen de la prÃĐsentation. La date de dÃĐpÃīt dâune prÃĐsentation admissible à un examen en continu serait la date à laquelle la prÃĐsentation est jugÃĐe administrativement complÃĻte par SantÃĐ Canada (câest-à -dire une fois que tous les ÃĐlÃĐments et formulaires requis pour le traitement sont remplis et ont ÃĐtÃĐ soumis). Cette date peut diffÃĐrer de la date de rÃĐception de la transaction initiale, si la soumission est considÃĐrÃĐe comme administrativement incomplÃĻte à ce moment. Une fois ÃĐtablie, la date de dÃĐpÃīt ne change pas, mÊme si, dans le cas dâun examen en continu, le fabricant fournit par la suite à SantÃĐ Canada les renseignements manquants.
Drogues dÃĐjà assujetties à un examen en continu selon la politique actuelle
En plus des options dâexamen en continu susmentionnÃĐes, conformÃĐment à la politique actuelle, les prÃĐsentations de mÃĐdicaments vÃĐtÃĐrinaires pour lesquelles le ministre a indiquÃĐ son intention de mener lâexamen avec un organisme de rÃĐglementation ÃĐtranger et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour lesquelles un changement a ÃĐtÃĐ apportÃĐ Ã la souche à laquelle se rapporte un vaccin antigrippal (par exemple les prÃĐsentations concernant les mises à jour annuelles des vaccins contre la grippe saisonniÃĻre) seraient ÃĐgalement admissibles à un examen en continu. Les vaccins antigrippaux admissibles à cette option dâexamen en continu seraient dÃĐfinis dans une liste incorporÃĐe par renvoi. Cette liste serait ÃĐlaborÃĐe, rÃĐvisÃĐe et maintenue conformÃĐment aux principes directeurs ÃĐnoncÃĐs dans la Politique dâincorporation par renvoi de SantÃĐ Canada. Afin de sâharmoniser avec la pratique actuelle, les exigences pour ces deux options dâexamen en continu seraient diffÃĐrentes. Par exemple, ces prÃĐsentations ne seraient pas assujetties à une ÃĐvaluation prÃĐalable à la prÃĐsentation.
à lâexception des examens en continu des prÃĐsentations oÃđ il y a eu un changement à la souche à laquelle un vaccin contre la grippe se rapporte, les modifications proposÃĐes pour les options dâexamen en continu dÃĐcrites ci-dessus permettraient ÃĐgalement au ministre, dans certaines conditions, dâannuler la prÃĐsentation. Par exemple, dans le cas des examens en continu de nouvelles drogues qui sont conformes aux conditions dÃĐcrites dans la section ci-dessus, lorsque le fabricant a omis, ou serait incapable, de fournir les renseignements manquants dans un dÃĐlai raisonnable aprÃĻs la date pertinente prÃĐcisÃĐe dans lâavis, lâexamen serait terminÃĐ et la prÃĐsentation serait considÃĐrÃĐe comme annulÃĐe par le fabricant. Si un fabricant choisit de dÃĐposer une nouvelle prÃĐsentation pour la mÊme drogue auprÃĻs de SantÃĐ Canada aprÃĻs une telle annulation, une nouvelle date de dÃĐpÃīt serait assignÃĐe une fois que la nouvelle prÃĐsentation est jugÃĐe administrativement complÃĻte.
Drogues dâurgence de santÃĐ publique
Enfin, les dispositions existantes du RAD concernant les activitÃĐs rÃĐglementÃĐes en vertu du titre 1A liÃĐes à la COVID-19 et lâautorisation de mise sur le marchÃĐ des drogues contre la COVID-19, y compris lâoption dâun examen en continu, seraient ÃĐlargies pour sâappliquer à toute drogue dâurgence de santÃĐ publique en relation avec une condition mentionnÃĐe dans la Liste dâaffections qui menacent la santÃĐ publique, qui serait incorporÃĐe par renvoi. Une drogue dâurgence de santÃĐ publique serait dÃĐfinie comme une nouvelle drogue dont le but et les conditions dâutilisation recommandÃĐes par le fabricant se rapportent à la COVID-19 ou à une affection dÃĐcrite dans la liste. Afin dâajouter des affections à la liste, le ministre devrait avoir des motifs raisonnables de croire que lâaffection prÃĐsente un risque important pour la santÃĐ publique au Canada ou en rÃĐsulte, et des mesures immÃĐdiates sont nÃĐcessaires pour faire face au risque. La proposition de dresser une liste qui serait incorporÃĐe par renvoi permettrait au ministre de rÃĐpondre à une urgence de santÃĐ publique dâune maniÃĻre opportune et souple. La liste serait ÃĐlaborÃĐe, examinÃĐe et tenue à jour conformÃĐment aux principes directeurs ÃĐnoncÃĐs dans la Politique dâincorporation par renvoi de SantÃĐ Canada.
à lâappui de lâÃĐlargissement de lâoption actuelle dâexamen en continu de la COVID-19 aux urgences de santÃĐ publique, les critÃĻres de prÃĐpositionnement dâune drogue seraient ÃĐgalement ÃĐlargis pour inclure une drogue dâurgence de santÃĐ publique afin de permettre une rÃĐponse à une affection mentionnÃĐe dans la liste. Le rÃĻglement serait ÃĐgalement modifiÃĐ pour permettre lâimportation seulement si le titulaire de la licence dâÃĐtablissement de produits pharmaceutiques est un importateur autorisÃĐ et que la drogue dâurgence de santÃĐ publique fait partie de la mÊme catÃĐgorie de drogue que celle autorisÃĐe par la licence.
Assurer la qualitÃĐ des drogues pendant la fabrication (modifications au RAD)
Les modifications proposÃĐes clarifieraient les attentes selon lesquelles une drogue doit Être fabriquÃĐe, emballÃĐe et ÃĐtiquetÃĐe, testÃĐe et entreposÃĐe, y compris entreposÃĐe pendant le transport, dâune maniÃĻre qui assure la qualitÃĐ de la drogue.
Cette exigence est censÃĐe sâappliquer à tous les fabricants, emballeurs/ÃĐtiqueteurs, testeurs et grossistes autorisÃĐs, ainsi quâaux importateurs et distributeurs canadiens. La nouvelle disposition C.02.012.1 vise à collaborer avec les autres exigences du titre 2 du RAD et ne modifie pas les attentes actuelles pour toutes les drogues.
Modernisation des exigences concernant les mÃĐdicaments biologiques (modifications au RAD)
La modernisation du titre 4 de la partie C du RAD envisage de supprimer les rÃĻglements dÃĐsuets spÃĐcifiques à certains produits biologiques et de les remplacer par des exigences plus gÃĐnÃĐrales qui reflÃĻtent les objectifs de sÃĐcuritÃĐ sous-jacents, tels quâils sâappliquent à la ligne directrice et à la pratique actuelles. Les dispositions quâon envisage dâabroger et de remplacer sont des dispositions propres à un produit allant de C.04.050 à C.04.683. Par consÃĐquent, les dÃĐfinitions de ÂŦ date de fabrication Âŧ et ÂŦ date de sortie Âŧ ne seraient plus nÃĐcessaires et nâapparaÃŪtraient pas dans le nouveau titre 4.
La modernisation proposÃĐe serait mise en Åuvre de façon à rÃĐduire au minimum lâincidence sur les fabricants de produits biologiques actuellement commercialisÃĐs en reconnaissant et en appuyant les pratiques actuelles. Plus prÃĐcisÃĐment, on ne sâattend pas à ce que les modifications proposÃĐes au RAD modifient les pratiques actuelles, y compris celles relatives à lâautorisation de mise en circulation de lots et aux ESP.
ContrÃīles sur la fabrication des produits biologiques
Les modifications proposÃĐes tiennent compte de la pratique actuelle et remplaceraient les exigences normatives concernant le matÃĐriel biologique de dÃĐpart et le matÃĐriel auxiliaire utilisÃĐs dans la fabrication de produits biologiques par des rÃĻglements plus souples et axÃĐs sur les rÃĐsultats qui maintiennent un niveau appropriÃĐ de surveillance en matiÃĻre dâinnocuitÃĐ. Les matÃĐriels dâorigine biologique ont ÃĐtÃĐ dÃĐfinis de maniÃĻre à inclure les matiÃĻres premiÃĻres biologiques, les matÃĐriels dâorigine biologique de dÃĐpart et les matÃĐriels auxiliaires. Les exigences relatives à la fabrication et à la collecte de produits biologiques ont ÃĐtÃĐ clarifiÃĐes afin de sâappliquer ÃĐgalement aux matÃĐriels dâorigine biologique.
Il est proposÃĐ de gÃĐnÃĐraliser les rÃĻglements existants qui contrÃīlent la contamination des produits biologiques afin de sâassurer que le personnel de production ne contribue pas au risque de contamination des matÃĐriels dâorigine biologique par des agents infectieux. Les modifications proposÃĐes supprimeraient certaines dispositions relatives aux exigences en matiÃĻre de personnel, dâentreposage et de transport, car elles sont dÃĐsuÃĻtes, et le MinistÃĻre se fonderait plutÃīt sur les rÃĻglements correspondants du titre 2 (Bonnes pratiques de fabrication) de la partie C du RAD.
Normes sur les produits biologiques
Les normes individuelles sur les produits biologiques, comme lâinsuline, qui sont prescrites au titre 4, sont dÃĐpassÃĐes par les changements scientifiques et ne sont pas actuellement utilisÃĐes par les fabricants ou SantÃĐ Canada. Lâintention est de remplacer ces normes prescrites et de continuer à ÃĐvaluer les spÃĐcifications fournies par le fabricant pendant lâexamen de la prÃĐsentation. De plus, les modifications proposÃĐes exigeraient que les prÃĐparations de rÃĐfÃĐrence utilisÃĐes pour ÃĐvaluer la puretÃĐ ou la puissance dâune drogue, selon le cas, soient suffisantes pour contrÃīler la qualitÃĐ du produit.
Mise en circulation de lots de produits biologiques
Les dispositions à lâappui du Programme de mise en circulation de lots seraient modifiÃĐes pour mieux appuyer une approche ÃĐchelonnÃĐe et fondÃĐe sur les risques, conformÃĐment à la Ligne directrice à lâintention des promoteurs : Programme dâautorisation de mise en circulation des lots de drogues visÃĐes à lâannexe D (produits biologiques) et la pratique actuelle.
Pour gÃĐrer les risques associÃĐs aux lots individuels, les modifications proposÃĐes permettraient au ministre de demander des renseignements, des ÃĐchantillons ou dâautres documents, au besoin, y compris pour effectuer des essais indÃĐpendants. Lorsquâune demande a ÃĐtÃĐ faite, personne ne serait autorisÃĐ Ã vendre une drogue de ce lot tant que le ministre ne lâaura pas avisÃĐe que le lot peut Être vendu.
Les modifications proposÃĐes officialiseraient ÃĐgalement dans la rÃĐglementation la pratique discrÃĐtionnaire actuelle de fournir des rapports annuels sur les produits biologiques. Ces rapports caractÃĐrisent la qualitÃĐ de la drogue et de ses ingrÃĐdients actifs, ainsi que la rÃĐgularitÃĐ de leurs procÃĐdÃĐs de fabrication et dâemballage au cours de la pÃĐriode ÃĐcoulÃĐe depuis le dernier rapport. Les exigences proposÃĐes comprendraient des rapports pÃĐriodiques sur la qualitÃĐ, sur une base annuelle ou plus longtemps, tel quâil est prÃĐcisÃĐ par le ministre.
Ãtiquetage de produits biologiques
Les modifications proposÃĐes mettraient à jour certaines exigences en matiÃĻre dâÃĐtiquetage pour les produits biologiques afin de les harmoniser avec les exigences du titre 1 de la partie C du RAD. Il sâagit notamment de la souplesse dâÃĐtiquetage des petits continents qui ont une ÃĐtiquette extÃĐrieure.
En plus des directives dâutilisation adÃĐquates, le rÃĻglement proposÃĐ exigerait sur lâÃĐtiquette une dÃĐclaration indiquant les conditions dâentreposage approuvÃĐes et des renseignements supplÃĐmentaires, au besoin, pour prÃĐvenir un prÃĐjudice à la santÃĐ du consommateur ou du patient (par exemple multidose, usage pÃĐdiatrique, groupe dâÃĒge, avertissement, etc.).
Les ÃĐtiquettes des produits biologiques devraient indiquer si la drogue provient directement dâune source humaine ou dâune source animale et devraient prÃĐciser lâespÃĻce dâorigine.
Les modifications proposÃĐes permettraient aux fabricants de produits biologiques entreposÃĐs par des ministÃĻres ou des organismes gouvernementaux pour Être utilisÃĐs dans des situations dâurgence, lorsquâon prÃĐvoit que la drogue sera entreposÃĐe pendant des pÃĐriodes prolongÃĐes et que le programme dâÃĐvaluation de la stabilitÃĐ Ã long terme est en cours, dâindiquer la date dâexpiration par dâautres moyens que par une dÃĐclaration sur lâÃĐtiquette de lâemballage. Les autres moyens dâindiquer la date dâexpiration devraient Être facilement accessibles à la personne qui administre la drogue et jugÃĐe acceptable par le ministre.
Toutes ces modifications proposÃĐes à lâÃĐtiquetage sont conformes aux pratiques actuelles et ne devraient donc pas avoir dâincidence sur les ÃĐtiquettes approuvÃĐes.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue (modifications au RAD)
ConformÃĐment à la pratique actuelle, la modification proposÃĐe à la disposition C.08.003.1 clarifierait le pouvoir du ministre dâexaminer des renseignements ou du matÃĐriel qui pourraient Être examinÃĐs au cas par cas en fonction des risques au cours de lâÃĐvaluation dâune prÃĐsentation de drogue par SantÃĐ Canada. Le ministre pourrait examiner des renseignements et du matÃĐriel :
- fournis au ministre par toute personne en vertu de la Loi;
- obtenus dâun immeuble ou dâun site oÃđ une drogue est fabriquÃĐe, emballÃĐe, ÃĐtiquetÃĐe ou analysÃĐe (par exemple ÃĐvaluations sur place pour les produits biologiques, inspection des BPF);
- obtenus dâune autoritÃĐ de rÃĐglementation ÃĐtrangÃĻre (par exemple rapports dâexamen ÃĐtrangers, rapports dâinspection ÃĐtrangers).
La disposition mise à jour nâa pas pour but de modifier lâobligation du fabricant de fournir suffisamment dâinformation à lâappui de la prÃĐsentation. Les dispositions relatives à la protection des donnÃĐes du RAD et le RÃĻglement sur les mÃĐdicaments brevetÃĐs (avis de conformitÃĐ) continueraient de sâappliquer, mÊme lorsque des renseignements sont pris en considÃĐration en vertu de la disposition C.08.003.1.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain (modifications au RAD)
Les modifications proposÃĐes obligeraient les fabricants à soumettre des donnÃĐes dâessais cliniques humains ventilÃĐes en sous-groupes de population pour appuyer lâinnocuitÃĐ et lâefficacitÃĐ de la prÃĐsentation de drogue nouvelle (ou un supplÃĐment), si les donnÃĐes dÃĐsagrÃĐgÃĐes ont dÃĐjà ÃĐtÃĐ soumises à la FDA ou à lâEMA. Il sâagit dâune ÃĐtape importante à mesure que SantÃĐ Canada va de lâavant avec une approche progressive en ce qui concerne les exigences supplÃĐmentaires en matiÃĻre de dÃĐsagrÃĐgation de donnÃĐes.
Normes (modifications au RAD)
Lâexemption rÃĐglementaire proposÃĐe concernant les normes exclurait les nouvelles drogues qui sont rÃĐglementÃĐes en vertu du titre 8 de la partie C du RAD, autres que celles de lâannexe C (produits pharmaceutiques radioactifs), dâavoir à respecter les normes les plus strictes de puretÃĐ et de puissance de toutes les pharmacopÃĐes de lâannexe B dans lesquelles se trouve lâingrÃĐdient actif ou la drogue, lorsque le fabricant revendique une norme dâun fabricant. En raison de leurs caractÃĐristiques intrinsÃĻques uniques et de leur profil de biodistribution sensible qui, en fin de compte, ont une incidence sur leur index thÃĐrapeutique et diagnostique, les drogues de lâannexe C continueraient dâÊtre tenues de respecter les normes les plus strictes en matiÃĻre de puretÃĐ et de puissance de toutes les pharmacopÃĐes de lâannexe B lorsquâune norme du fabricant est revendiquÃĐe.
Les drogues qui ne sont pas de nouvelles drogues continueraient dâÊtre assujetties aux exigences actuelles.
Pour les drogues rÃĐglementÃĐes uniquement en vertu du titre 1 et celles rÃĐglementÃĐes en vertu du titre 8 qui ne sont pas ÃĐgalement rÃĐglementÃĐes en vertu des titres 3 et 4 (câest-à -dire les produits pharmaceutiques radioactifs et biologiques), les modifications proposÃĐes ÃĐlimineraient lâexigence selon laquelle la norme utilisÃĐe pour la drogue doit Être indiquÃĐe sur lâÃĐtiquette de lâemballage.
EntrÃĐe en vigueur
Les modifications rÃĐglementaires proposÃĐes suivantes, qui accordent la prioritÃĐ Ã la rÃĐduction du fardeau pour lâindustrie et font en sorte que SantÃĐ Canada soit bien placÃĐ pour faire face à toute urgence de santÃĐ publique future, entreraient en vigueur au moment de lâenregistrement :
- les modifications relatives aux drogues dâurgence de santÃĐ publique (y compris les conditions, les examens en continu et le prÃĐpositionnement);
- assurer la qualitÃĐ des drogues pendant la fabrication;
- la modernisation des exigences concernant les produits biologiques;
- les renseignements à lâappui de lâexamen des prÃĐsentations de drogue;
- les donnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogues nouvelles pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain;
- les normes.
Les modifications rÃĐglementaires proposÃĐes suivantes, qui nÃĐcessitent un dÃĐlai supplÃĐmentaire pour Être mises en Åuvre, entreraient en vigueur un an aprÃĻs lâenregistrement :
- les conditions (pour toutes les drogues et les instruments mÃĐdicaux de classe II, III et IV);
- les plans de gestion des risques;
- les examens en continu (pour toutes les drogues sauf les drogues dâurgence de santÃĐ publique).
Les modifications rÃĐglementaires proposÃĐes suivantes entreraient en vigueur à un moment fixÃĐ dans une modification rÃĐglementaire future une fois que le ministÃĻre aura dÃĐterminÃĐ que les dispositions propres à la COVID-19 ne sont plus nÃĐcessaires :
- modifier la dÃĐfinition de drogue dâurgence de santÃĐ publique pour ne plus inclure une drogue nouvelle dont lâusage ou les conditions dâemploi recommandÃĐs par le fabricant se rapportent à la COVID-19;
- abroger dâautres dispositions spÃĐcifiques à la COVID-19.
Ãlaboration de la rÃĐglementation
Consultation
Consultations antÃĐrieures
Les lignes directrices et les politiques existantes, qui constituent la base de cette proposition, ont fait lâobjet de consultations avant leur mise en Åuvre et sont maintenant de pratique courante. Par exemple, lâapproche stratÃĐgique de SantÃĐ Canada à lâÃĐgard des PGR a ÃĐtÃĐ ÃĐtablie en fonction des lignes directrices E2E de la ConfÃĐrence internationale sur lâharmonisation des exigences techniques relatives à lâhomologation des produits pharmaceutiques pour usage humain (PDF, disponible en anglais seulement). Ces lignes directrices ÃĐtaient fondÃĐes sur les commentaires reçus de lâindustrie pharmaceutique internationale. Avant la mise en Åuvre des lignes directrices finales de SantÃĐ Canada sur les PGR, qui ont ÃĐtÃĐ bien reçues par lâindustrie, une importante consultation a ÃĐtÃĐ menÃĐe pour assurer lâharmonisation avec les lignes directrices internationales qui ont ÃĐtÃĐ mises en Åuvre par dâautres administrations.
SantÃĐ Canada a ÃĐgalement entendu les intervenants sur la nÃĐcessitÃĐ dâune souplesse rÃĐglementaire au cours du processus pour lâexamen de la rÃĐglementation du secteur des sciences de la santÃĐ et des sciences biologiques de 2018.
De plus, des consultations ont eu lieu avant de prendre les arrÊtÃĐs dâurgence et les rÃĻglements provisoires portant sur la COVID-19. Les rÃĐsultats de ces consultations sont rÃĐsumÃĐs dans un rapport Ce qui a ÃĐtÃĐ entendu affichÃĐ en ligne, ainsi que dans les notes explicatives sur les arrÊtÃĐs dâurgence concernant les instruments mÃĐdicaux et le rÃĐsumÃĐ de lâÃĐtude dâimpact de la rÃĐglementation accompagnant le rÃĻglement qui a transformÃĐ lâarrÊtÃĐ dâurgence concernant les mÃĐdicaments en rÃĻglement. Les intervenants nâont soulevÃĐ aucune prÃĐoccupation quant à la capacitÃĐ du ministre dâimposer des conditions sur les drogues contre la COVID-19 et les instruments mÃĐdicaux destinÃĐs à Être utilisÃĐs à lâÃĐgard de la COVID-19. Les intervenants ÃĐtaient ÃĐgalement favorables à lâutilisation dâexamens en continu, et un intervenant a suggÃĐrÃĐ que les examens en continu soient ÃĐlargis à dâautres drogues dâimportance vitale.
Au fil des annÃĐes, les fabricants ont demandÃĐ de faire preuve de souplesse dans les exigences en matiÃĻre dâÃĐtiquetage relatives aux normes pour les drogues. De plus, lorsquâils revendiquent la norme dâun fabricant, les fabricants ont indiquÃĐ que les exigences actuelles sont trop restrictives en ce qui concerne la puretÃĐ et la puissance dâune drogue.
Les intervenants ont exprimÃĐ leur appui à la modernisation des exigences relatives aux produits biologiques au fil des ans. Depuis plus de 10 ans, le ministÃĻre a effectuÃĐ des analyses techniques, scientifiques et stratÃĐgiques importantes du titre 4 et de lâannexe D. Tout au long de cette analyse, les intervenants de lâindustrie ont ÃĐtÃĐ consultÃĐs et ont appuyÃĐ les efforts de modernisation de SantÃĐ Canada.
Avis dâintention
Les commentaires des intervenants sur certaines des composantes ont ÃĐtÃĐ demandÃĐs à la suite de la publication dâun avis dâintention de 90 jours dans la Partie I de la Gazette du Canada. La consultation a eu lieu du 31 juillet 2021 au 28 octobre 2021. Pendant cette pÃĐriode, on a ÃĐgalement demandÃĐ aux intervenants de lâindustrie de remplir un sondage sur les coÃŧts-avantages. Des rÃĐunions ont ÃĐgalement eu lieu avec des organismes dâÃĐvaluation des technologies de la santÃĐ (ETS) et des partenaires du systÃĻme de santÃĐ comme les provinces et les territoires. Ces rÃĐunions ont servi à donner un aperçu de lâavis dâintention et à rÃĐpondre aux questions des intervenants au sujet des modifications proposÃĐes au RAD et au RIM.
RÃĐponse
SantÃĐ Canada a reçu 25 rÃĐponses de la part dâentreprises pharmaceutiques et dâinstruments mÃĐdicaux, dâassociations de lâindustrie, dâorganismes dâETS, de provinces et de territoires, dâassociations reprÃĐsentant des professionnels de la santÃĐ et des particuliers, ainsi que 32 sondages sur les coÃŧts-avantages rÃĐalisÃĐs auprÃĻs dâintervenants de lâindustrie. Les commentaires reçus ont ÃĐtÃĐ utilisÃĐs pour peaufiner les modifications proposÃĐes incluses dans cette proposition.
Les intervenants ÃĐtaient gÃĐnÃĐralement en faveur des modifications proposÃĐes; toutefois, ils ont demandÃĐ que des dÃĐtails supplÃĐmentaires concernant la mise en Åuvre soient fournis. Ils ont notamment exprimÃĐ leur soutien aux efforts de SantÃĐ Canada pour accroÃŪtre la souplesse rÃĐglementaire, rÃĐduire les irritants connus, amÃĐliorer lâaccÃĻs aux produits thÃĐrapeutiques et amÃĐliorer la surveillance aprÃĻs la mise en marchÃĐ tout au long du cycle de vie dâune drogue ou dâun instrument mÃĐdical.
Conditions — Drogues et instruments mÃĐdicaux (modifications au RAD et au RIM)
En ce qui concerne les conditions, certains intervenants de lâindustrie ont recommandÃĐ que les conditions relatives aux drogues et aux instruments mÃĐdicaux ne soient utilisÃĐes que dans des circonstances exceptionnelles. Les organismes dâETS voulaient mieux comprendre les rÃĐpercussions des conditions sur leurs processus dâexamen. Les rÃĐpondants ont ÃĐgalement indiquÃĐ que les conditions devraient Être harmonisÃĐes à lâÃĐchelle internationale afin de rÃĐduire le fardeau et, par consÃĐquent, les coÃŧts pour lâindustrie, et quâune possibilitÃĐ dâÊtre entendu devrait Être offerte au fabricant avant que les conditions ne soient appliquÃĐes à lâidentification numÃĐrique dâune drogue. Une association reprÃĐsentant les pharmaciens a indiquÃĐ que les conditions des drogues ne devraient pas se traduire par des exigences prÃĐalables à la mise en marchÃĐ moins rigoureuses et quâil faudrait prendre soin de ne pas avoir dâincidence sur la pratique des professionnels de la santÃĐ. Un intervenant du secteur des instruments mÃĐdicaux a remis en question le besoin de conditions pour les instruments de classe II (câest-à -dire les instruments à fiable risque). Un intervenant de lâindustrie a indiquÃĐ quâil serait judicieux dâharmoniser les exigences des conditions pour les drogues et les instruments mÃĐdicaux, ÃĐtant donnÃĐ quâil existe des produits de santÃĐ Ã la frontiÃĻre entre les drogues et les instruments mÃĐdicaux. Pour rÃĐpondre à ces prÃĐoccupations, les modifications proposÃĐes au RAD et au RIM ÃĐnoncent des considÃĐrations dont le ministre devrait tenir compte avant dâimposer des conditions. Les conditions ne seraient pas utilisÃĐes comme mÃĐcanisme pour remplacer les exigences prÃĐalables à la mise en marchÃĐ.
PGR (modifications au RAD)
Certains intervenants ont exprimÃĐ des prÃĐoccupations au sujet de fausses dÃĐclarations si SantÃĐ Canada devait rÃĐdiger et traduire des rÃĐsumÃĐs de leurs PGR. En rÃĐponse à cette prÃĐoccupation, les modifications rÃĐglementaires proposÃĐes exigeraient que les fabricants incluent des rÃĐsumÃĐs de leurs PGR dans les deux langues officielles au moment du dÃĐpÃīt de la prÃĐsentation pour examen par SantÃĐ Canada.
Les intervenants ont exprimÃĐ une volontÃĐ de maintenir certaines pratiques opÃĐrationnelles existantes, comme la capacitÃĐ de nÃĐgocier le contenu des PGR pendant le processus dâexamen de prÃĐsentations de drogues. De plus, les intervenants ont indiquÃĐ que les PGR ne devraient pas avoir dâincidence sur la pratique des professionnels de la santÃĐ (par exemple les programmes de distribution contrÃīlÃĐe qui auraient une incidence sur la capacitÃĐ dâun pharmacien à prescrire une drogue).
Les organismes dâETS ont indiquÃĐ quâils aimeraient obtenir des dÃĐtails supplÃĐmentaires sur les opÃĐrations et la mise en Åuvre de la façon dont les PGR seraient examinÃĐs par SantÃĐ Canada en raison dâune incidence prÃĐvue sur leurs processus dâexamen.
Examens en continu (modifications au RAD)
Les organismes dâETS voulaient aussi mieux comprendre comment les examens en continu seraient mis en Åuvre et lâeffet que ces modifications proposÃĐes pourraient avoir sur leur processus dâexamen. De plus, les intervenants de lâindustrie se sont gÃĐnÃĐralement prononcÃĐs en faveur de lâoption des examens en continu; toutefois, ils ont demandÃĐ des prÃĐcisions supplÃĐmentaires sur les types de drogues qui seraient admissibles à un examen en continu et sur la façon dont ces modifications seraient mises en Åuvre. Les intervenants ont ÃĐgalement fait part de leurs attentes quant aux avantages dâun examen en continu qui devraient Être ÃĐvidents par rapport aux autres options dâÃĐvaluations prioritaires des prÃĐsentations.
Modernisation des exigences concernant les produits biologiques (modifications au RAD)
En ce qui concerne la modernisation des dispositions relatives aux produits biologiques, les intervenants ont indiquÃĐ que lâapproche rÃĐglementaire finale pour les analyses de mise en circulation de lots devrait Être fondÃĐe sur les risques et souple. SantÃĐ Canada utilise actuellement lâapproche ÃĐchelonnÃĐe et fondÃĐe sur les risques dÃĐcrite dans la Ligne directrice à lâintention des promoteurs : Programme dâautorisation de mise en circulation des lots de drogues visÃĐes à lâannexe D (produits biologiques). Les modifications rÃĐglementaires proposÃĐes permettraient lâutilisation continue dâune approche axÃĐe sur les risques.
Les intervenants ÃĐtaient en faveur de la modernisation des exigences en matiÃĻre dâÃĐtiquetage des produits biologiques, mais ils ont suggÃĐrÃĐ que des considÃĐrations dâespacement soient prises pour les petites ÃĐtiquettes, que lâacceptation dâÃĐtiquettes universelles et de codes lisibles par machine soit prise en considÃĐration et que tout changement, comme lâexigence proposÃĐe dâindiquer lâespÃĻce dâorigine, ne devrait pas avoir dâincidence sur la capacitÃĐ des professionnels de la santÃĐ de prescrire certaines drogues. Plusieurs intervenants ont dÃĐclarÃĐ que si dâautres moyens dâindiquer la date dâexpiration des drogues entreposÃĐes ÃĐtaient autorisÃĐs, lâexigence devrait faire en sorte que la date soit rapidement et facilement disponible aux professionnels et aux patients qui utilisent la drogue. Les modifications rÃĐglementaires proposÃĐes pour lâÃĐtiquetage des produits biologiques dans de petits contenants offriraient aux fabricants la souplesse dÃĐjà offerte au titre 1 pour les autres drogues. à lâheure actuelle, il nây a aucune restriction à lâutilisation dâÃĐtiquettes universelles ou de codes lisibles par machine, pourvu quâils respectent les exigences de la lÃĐgislation canadienne. SantÃĐ Canada continue dâappuyer la capacitÃĐ des professionnels de la santÃĐ Ã prescrire des drogues, notamment en veillant à ce que lâinformation sur une drogue soit claire et accessible.
Bien que les ESP soient une pratique courante, certains intervenants de lâindustrie ont indiquÃĐ que cela est une exigence propre au Canada et quâelle reprÃĐsente donc un fardeau supplÃĐmentaire. Certains intervenants ont suggÃĐrÃĐ que SantÃĐ Canada ÃĐlimine cette pratique au profit de lâutilisation dâinformation provenant dâautres sources, comme les inspections des BPF. Dâautres intervenants ont indiquÃĐ que les ESP devraient Être flexibles et axÃĐes sur le risque, ou davantage harmonisÃĐes avec les inspections propres aux produits comme celles effectuÃĐes par lâEMA ou la FDA. Les inspections des BPF diffÃĻrent des ESP en ce sens que les inspections des BPF ne sont pas propres au produit. Les ESP sont un outil essentiel utilisÃĐ par le ministÃĻre dâune maniÃĻre axÃĐe sur les risques pour un sous-ensemble de prÃĐsentations de drogue. Il sâagit dâune activitÃĐ liÃĐe aux prÃĐsentations, qui consiste à ÃĐvaluer le processus de fabrication du produit, à valider lâinformation fournie dans la prÃĐsentation de drogue et à ÃĐvaluer le processus de fabrication mis en Åuvre au site. SantÃĐ Canada applique actuellement une approche fondÃĐe sur le risque pour dÃĐcider si des renseignements supplÃĐmentaires obtenus des ESP concernant la mise en Åuvre du processus de fabrication dans les installations proposÃĐes sont nÃĐcessaires pour appuyer la prise de dÃĐcisions concernant les prÃĐsentations de drogue nouvelle ou les supplÃĐments à une prÃĐsentation de drogue nouvelle pour les produits biologiques. Lâapproche fondÃĐe sur le risque tient compte de facteurs comme la complexitÃĐ ou la nouveautÃĐ du processus de fabrication, la connaissance du site par SantÃĐ Canada et lâexpÃĐrience du fabricant dans la production dâune drogue sÃĐcuritaire et uniforme. La modification rÃĐglementaire proposÃĐe assurerait la transparence en ce qui concerne lâinclusion de lâinformation obtenue directement ou indirectement à partir des sites de fabrication dans le processus dÃĐcisionnel pour lâautorisation de mise en marchÃĐ.
Hors de portÃĐe
SantÃĐ Canada a ÃĐgalement reçu un certain nombre de commentaires qui nâentraient pas dans la portÃĐe de cette proposition rÃĐglementaire. Ceux-ci comprenaient les commentaires sur le processus dâautorisation des essais cliniques, les souplesses liÃĐes à lâÃĐtiquetage et à lâaccÃĻs au marchÃĐ des mÃĐdicaments gÃĐnÃĐriques, les examens en continu des licences dâÃĐtablissement de produits pharmaceutiques et des instruments mÃĐdicaux, et les modifications aux frais dâautorisation des mÃĐdicaments vÃĐtÃĐrinaires afin de faciliter lâaccÃĻs au marchÃĐ et dâÊtre concurrentiel à lâÃĐchelle mondiale.
Obligations relatives aux traitÃĐs modernes et consultation et mobilisation des Autochtones
Comme lâexige la Directive du Cabinet sur lâapproche fÃĐdÃĐrale pour la mise en Åuvre des traitÃĐs modernes, le projet a fait lâobjet dâune ÃĐvaluation dÃĐtaillÃĐe des rÃĐpercussions des traitÃĐs modernes. LâÃĐvaluation nâa cernÃĐ aucune rÃĐpercussion sur ou obligation envers les traitÃĐs modernes.
Choix de lâinstrument
SantÃĐ Canada a examinÃĐ les options rÃĐglementaires et non rÃĐglementaires suivantes.
Option 1 : Ne pas introduire de modifications rÃĐglementaires et maintenir le statu quo
Sans les modifications proposÃĐes au RAD et au RIM, le ministÃĻre nâatteindrait pas son objectif de fournir un pouvoir rÃĐglementaire clair pour les souplesses, dont certaines sont actuellement en place par le biais de politiques, pour suivre le rythme de lâinnovation et faciliter lâaccÃĻs à des traitements de pointe et à des thÃĐrapies prometteuses, tout en continuant de sâassurer que les drogues autorisÃĐes et les instruments mÃĐdicaux homologuÃĐs sont sÃŧrs, efficaces et assujettis à une surveillance appropriÃĐe, conformÃĐment à son engagement pris dans la Feuille de route du secteur de la santÃĐ et des sciences biologiques. Lâabsence de modifications visant à codifier ces souplesses donne lieu à un manque de certitude pour les fabricants quant au fait que les souplesses seraient disponibles et appliquÃĐes de façon uniforme.
De plus, des engagements juridiquement contraignants pour la gestion des risques et des incertitudes au moyen des conditions continueraient de sâappliquer seulement aux opioÃŊdes et aux drogues contre la COVID-19. En vertu du RIM, les pouvoirs des conditions seraient limitÃĐs aux essais pour les instruments de classe II à IV, tandis que les conditions gÃĐnÃĐrales sâappliqueraient uniquement aux instruments mÃĐdicaux destinÃĐs à Être utilisÃĐs à lâÃĐgard de la COVID-19 autorisÃĐs en vertu de lâarrÊtÃĐ dâurgence actuel.
Lâoption de maintenir le statu quo ne garantit pas quâil existe des fondements juridiques en place pour mettre en Åuvre un cadre sur les drogues et les instruments mÃĐdicaux qui surveille efficacement les drogues et les instruments mÃĐdicaux rÃĐglementÃĐs tout au long de leur cycle de vie.
Option 2 : Proposer des modifications rÃĐglementaires visant à introduire des ÃĐlÃĐments de modernisation au cadre rÃĐglementaire pour les drogues et les instruments mÃĐdicaux
Il sâagit de lâoption prÃĐfÃĐrÃĐe, car elle garantirait que le ministre dispose des outils appropriÃĐs pour surveiller lâinnocuitÃĐ et lâefficacitÃĐ des drogues et des instruments mÃĐdicaux au moyen de conditions exÃĐcutoires. Le Canada passerait dâun cadre juridique oÃđ SantÃĐ Canada rÃĐagit passivement aux problÃĻmes à un cadre oÃđ les risques sont attÃĐnuÃĐs de façon proactive au moyen dâexigences rÃĐglementaires comme les conditions et les PGR. Elle favoriserait ÃĐgalement lâaccÃĻs rapide des Canadiens aux drogues essentielles en incluant une option pour un examen en continu.
Cette option permettait au ministÃĻre de commencer à respecter les engagements pris dans la Feuille de route du secteur de la santÃĐ et des sciences biologiques, de rÃĐduire le fardeau sur lâindustrie et de mettre ses rÃĻglements à jour avec la pratique actuelle afin quâils soient clairs pour les intervenants.
Analyse de la rÃĐglementation
Avantages et coÃŧts
Lâanalyse coÃŧts-avantages (ACA) vise à quantifier les avantages et les coÃŧts du projet de RÃĻglement modifiant certains rÃĻglements pris en application de la Loi sur les aliments et drogues (homologation agile).
Le rapport complet sur lâACA est disponible sur demande.
ÃnoncÃĐ des coÃŧts et des avantages
Les coÃŧts supplÃĐmentaires pour lâindustrie sont estimÃĐs à 158 millions de dollars (VA) sur une pÃĐriode de 10 ans. Il devrait coÃŧter au gouvernement au cours de la mÊme pÃĐriode 26 millions de dollars (VA) pour examiner et gÃĐrer les conditions et les examens en continu. Par consÃĐquent, le coÃŧt total prÃĐvu du rÃĻglement proposÃĐ en VA est de 184 millions de dollars sur une pÃĐriode de 10 ans, actualisÃĐ Ã 7 %, ou une moyenne annualisÃĐe dâenviron 26 millions de dollars.
Les avantages quantifiÃĐs comprennent des ventes estimÃĐes à 70 millions de dollars en VA provenant de lâobtention dâune autorisation de marchÃĐ deux mois plus tÃīt que dans le cadre du statu quo. Les Canadiens devraient bÃĐnÃĐficier dâune amÃĐlioration de leur qualitÃĐ de vie grÃĒce à une VA de 302 millions de dollars. De plus, lâÃĐlimination des exigences rÃĐglementaires relatives aux normes devrait profiter à lâindustrie de 68 millions de dollars (VA). Lâavantage total prÃĐvu se chiffrerait à 440 millions de dollars sur une pÃĐriode de 10 ans, actualisÃĐ Ã 7 %, soit une moyenne annualisÃĐe dâenviron 63 millions de dollars (VA).
- Nombre dâannÃĐes : 10 (de 2024 Ã 2033)
- AnnÃĐe du niveau de prix : 2021
- AnnÃĐe de rÃĐfÃĐrence de la VA : 2024
- Taux dâactualisation : 7 %
| Intervenant touchÃĐ | Description de lâavantage | AnnÃĐe 1 | AnnÃĐe 2 | AnnÃĐe 3 | DerniÃĻre annÃĐe | Total (VA) | Valeur annualisÃĐe |
|---|---|---|---|---|---|---|---|
| Industrie | Deux mois de ventes | 0 $ | 10 710 000 $ | 10 710 000 $ | 10 710 000 $ | 69 778 137 $ | 9 934 837 $ |
| Industrie | Normes | 9 112 500 $ | 9 112 500 $ | 9 112 500 $ | 9 112 500 $ | 68 482 554 $ | 9 750 375 $ |
| Canadiens | Deux mois dâAVAQ note a du tableau b1 | 0 $ | 44 720 676 $ | 45 167 883 $ | 48 426 084 $ | 301 944 819 $ | 42 990 149 $ |
| Tous les intervenants | Total des avantages | 9 112 500 $ | 64 543 176 $ | 64 990 383 $ | 68 248 584 $ | 440 205 510 $ | 62 675 361 $ |
Note(s) du tableau b1
|
|||||||
| Intervenant touchÃĐ | Description du coÃŧt | AnnÃĐe 1 | AnnÃĐe 2 | AnnÃĐe 3 | DerniÃĻre annÃĐe | Total (VA) | Valeur annualisÃĐe |
|---|---|---|---|---|---|---|---|
| Gouvernement | Conditions | 0 $ | 3 145 964 $ | 3 145 964 $ | 3 145 964 $ | 20 496 684 $ | 2 918 267 $ |
| Gouvernement | Examens en continu | 131 888 $ | 635 895 $ | 635 895 $ | 635 895 $ | 4 274 892 $ | 608 648 $ |
| Gouvernement | Solutions de TI | 1 028 207 $ | 65 887 $ | 65 887 $ | 65 887 $ | $1 457 476 $ | 207 512 $ |
| Industrie | Conditions — MÃĐdicaments | 0 $ | 7 293 333 $ | 14 586 667 $ | 21 880 000 $ | 122 550 603 $ | 17 448 449 $ |
| Industrie | Conditions — Instruments mÃĐdicaux | 0 $ | 1 090 000 $ | 2 180 000 $ | 3 270 000 $ | 18 315 378 $ | 2 607 698 $ |
| Industrie | PGR | 0 $ | 2 289 600 $ | 2 289 600 $ | 2 289 600 $ | 14 917 276 $ | 2 123 884 $ |
| Industrie | Examens en continu | 0 $ | 360 000 $ | 360 000 $ | 360 000 $ | 2 345 484 $ | 333 944 $ |
| Tous les intervenants | CoÃŧts totaux | 1 160 095 $ | 14 880 679 $ | 23 264 012 $ | 31 647 346 $ | 184 357 792 $ | 26 248 402 $ |
| RÃĐpercussions | AnnÃĐe 1 | AnnÃĐe 2 | AnnÃĐe 3 | DerniÃĻre annÃĐe | Total (VA) | Valeur annualisÃĐe |
|---|---|---|---|---|---|---|
| Total des avantages | 9 112 500 $ | 64 543 176 $ | 64 990 383 $ | 68 248 584 $ | 440 205 510 $ | 62 675 361 $ |
| CoÃŧts totaux | 1 160 095 $ | 14 880 679 $ | 23 264 012 $ | 31 647 346 $ | 184 357 792 $ | 26 248 402 $ |
| IMPACT NET | 7 952 405 $ | 49 662 497 $ | 41 726 370 $ | 36 601 238 $ | 255 847 718 $ | 36 426 959 $ |
ïŧŋEn ce qui concerne les avantages qualitatifs, on sâattend à ce que les modifications proposÃĐes apportent une clartÃĐ Ã lâindustrie et à lâorganisme de rÃĐglementation, ce qui permettra une plus grande souplesse pour faire face aux innovations futures. De plus, elles contribueraient à lâapproche du cycle de vie de la rÃĐglementation des produits thÃĐrapeutiques nÃĐcessaires pour protÃĐger les Canadiens contre les effets nocifs des produits thÃĐrapeutiques quâils utilisent tout en leur offrant les avantages thÃĐrapeutiques. Le rÃĐsultat dâune approche du cycle de vie devrait Être une rÃĐduction des effets indÃĐsirables et des incidents liÃĐs aux matÃĐriels mÃĐdicaux.
ScÃĐnario de rÃĐfÃĐrence
Le niveau de rÃĐfÃĐrence reflÃĻte les processus opÃĐrationnels et dâexamen actuels et sert de base au calcul de tout coÃŧt supplÃĐmentaire.
Avant dâadopter la Loi de Vanessa (2014), SantÃĐ Canada avait des options limitÃĐes lorsquâun problÃĻme grave dâinnocuitÃĐ se posait concernant un produit commercialisÃĐ. Le ministre avait la capacitÃĐ dâimposer des conditions touchant les essais pour les homologations dâinstruments mÃĐdicaux, mais cela nâÃĐtait pas le cas pour les autres produits. SantÃĐ Canada pouvait ÃĐgalement conserver un produit sur le marchÃĐ tout en ÃĐmettant un avertissement de sÃĐcuritÃĐ et en nÃĐgociant possiblement un changement dâÃĐtiquette avec le fabricant. Autrement, le produit aurait ÃĐtÃĐ retirÃĐ du marchÃĐ, ce qui pourrait priver les Canadiens de produits commercialisÃĐs pouvant sauver des vies. Les pouvoirs sur le cycle de vie instaurÃĐs par la Loi de Vanessa au niveau de la Loi ont amÃĐliorÃĐ les options rÃĐglementaires pour maintenir les produits sur le marchÃĐ tout en amÃĐliorant la capacitÃĐ de SantÃĐ Canada à rÃĐgler les problÃĻmes de sÃĐcuritÃĐ.
Ces pouvoirs ont aidÃĐ SantÃĐ Canada à adopter une approche de la rÃĐglementation axÃĐe sur le cycle de vie des produits; toutefois, un certain nombre de politiques de SantÃĐ Canada sont encore exercÃĐes au moyen de directives.
Conditions
Le ministre a actuellement le pouvoir dâimposer des conditions relatives aux essais sur les homologations dâinstruments mÃĐdicaux de classe II à IV et de modifier les conditions pour tenir compte de tout nouveau dÃĐveloppement relatif à lâinstrument. à lâheure actuelle, les rÃĻglements permettent ÃĐgalement au ministre dâimposer des conditions sur lâidentification numÃĐrique dâune drogue pour les opioÃŊdes et les drogues dÃĐsignÃĐes contre la COVID-19. En vertu de la politique sur les AC-C de SantÃĐ Canada, les fabricants acceptent de se conformer aux conditions visant à confirmer lâefficacitÃĐ des nouvelles drogues pour usage humain avec des preuves cliniques prometteuses pour traiter des affections graves ou mettant en danger la vie. Les conditions de la politique sur les AC-C sont semblables à celles des autres administrations; toutefois, elles reposent sur la conformitÃĐ volontaire et ne sont pas exÃĐcutoires. La politique sur les AC-C nâa pas ÃĐtÃĐ appliquÃĐe aux mÃĐdicaments vÃĐtÃĐrinaires et des conditions nâont pas ÃĐtÃĐ imposÃĐes aux identifications numÃĐriques dâune drogue des mÃĐdicaments vÃĐtÃĐrinaires.
Plans de gestion des risques
à lâheure actuelle, les fabricants se conforment volontairement aux directives sur la soumission des PGR. Habituellement, les PGR sont prÃĐsentÃĐs dans le mÊme format que celui exigÃĐ par lâEMA. SantÃĐ Canada engage certains coÃŧts liÃĐs aux nÃĐgociations pendant le processus dâexamen des prÃĐsentations pour harmoniser le contenu des PGR avec les recommandations dans les lignes directrices sur le contenu propre au Canada. Ãtant donnÃĐ que les PGR ne sont pas une exigence de la rÃĐglementation, le ministre a peu de pouvoir pour obliger un fabricant à soumettre un PGR complet en temps opportun, ce qui entraÃŪne un temps de nÃĐgociation avec le fabricant.
Examens en continu
à lâheure actuelle, le RAD envisage explicitement de procÃĐder à des examens en continu pour les drogues dÃĐsignÃĐes contre la COVID-19. Des examens en continu sont ÃĐgalement disponibles pour les mÃĐdicaments vÃĐtÃĐrinaires pour lesquels le ministre a indiquÃĐ son intention de mener lâexamen en collaboration avec un organisme de rÃĐglementation ÃĐtranger et pour les mises à jour annuelles des vaccins contre la grippe saisonniÃĻre.
ScÃĐnario rÃĐglementaire
Les modifications rÃĐglementaires sont dÃĐcrites sous chaque ÃĐlÃĐment proposÃĐ ci-dessous.
CoÃŧts pour lâindustrie
Conditions pour les drogues
En vertu du rÃĻglement proposÃĐ, des conditions pourraient Être imposÃĐes en tout temps, mais il est prÃĐvu quâelles ne seraient pas imposÃĐes à chaque drogue autorisÃĐe. Elles seraient utilisÃĐes pour gÃĐrer les incertitudes liÃĐes aux avantages et/ou aux risques (par exemple demande dâÃĐtudes cliniques confirmatives, dâÃĐtudes de qualitÃĐ et/ou dâefficacitÃĐ).
ConformÃĐment aux critÃĻres de la politique sur les AC-C, des conditions sont actuellement appliquÃĐes aux nouvelles drogues pour usage humain avec des preuves cliniques prometteuses pour traiter des affections graves ou mettant en danger la vie. Les coÃŧts et avantages supplÃĐmentaires seraient mesurÃĐs en fonction de la pratique actuelle de la politique sur les AC-C.
La politique sur les AC-C ne sâapplique pas aux mÃĐdicaments vÃĐtÃĐrinaires. Par consÃĐquent, à ce jour, aucune AC-C ni aucune condition nâa ÃĐtÃĐ imposÃĐe aux mÃĐdicaments vÃĐtÃĐrinaires. Toutefois, les modifications rÃĐglementaires proposÃĐes dÃĐfinissant les conditions permettraient aux fabricants de mÃĐdicaments vÃĐtÃĐrinaires dâÊtre assujettis à ce pouvoir.
En moyenne chaque annÃĐe, SantÃĐ Canada ÃĐmet sept AC-C. En fonction de lâanalyse interne et de lâÃĐlargissement de la portÃĐe de la proposition, on prÃĐvoit une augmentation de 10 conditions ÃĐmises chaque annÃĐe. Dans le cadre dâune enquÊte sur lâÃĐtablissement des coÃŧts envoyÃĐ Ã lâindustrie (enquÊte sur lâACA) menÃĐe entre juillet et octobre 2021, les intervenants ont indiquÃĐ un large ÃĐventail de coÃŧts des conditions, à la fois en fonction de leur expÃĐrience avec les AC-C et les conditions dans dâautres administrations et des coÃŧts prÃĐvus. Les coÃŧts dÃĐclarÃĐs varient de 4 000 $ (prÃĐsentation des rÃĐsultats dâune ÃĐtude) à 6,2 millions de dollars (rÃĐalisation dâune ÃĐtude clinique). DâaprÃĻs les rÃĐponses de ceux qui ont eu de lâexpÃĐrience avec les AC-C et les conditions, les coÃŧts moyens des conditions sont estimÃĐs à 2,2 millions de dollars. Ces coÃŧts comprendraient la production de rapports dâÃĐtudes cliniques pour les ÃĐtudes en cours, la surveillance et la fourniture de toutes les mises à jour sur les changements dans les maladies et la fourniture de renseignements supplÃĐmentaires.
On suppose que des conditions se rÃĐgleraient dâici trois ans, comme lâont indiquÃĐ certains intervenants, et que leur coÃŧt serait rÃĐparti ÃĐgalement sur cette pÃĐriode.
Le coÃŧt estimÃĐ des conditions pour lâindustrie au cours de la premiÃĻre annÃĐe de mise en Åuvre (deuxiÃĻme annÃĐe du rÃĻglement) sont de 7 293 333 $, de 14 586 667 $ pour la deuxiÃĻme annÃĐe et de 21 880 000 $ pour chaque annÃĐe suivante. La VA est dâenviron 122 550 603 $ sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Conditions pour les instruments mÃĐdicaux
En vertu du prÃĐsent RIM, les fabricants sont tenus de se conformer aux conditions qui sont imposÃĐes sur les homologations dâinstruments mÃĐdicaux de classe II à IV, et la capacitÃĐ du ministre dâimposer des conditions est limitÃĐe aux exigences dâessai. Le rÃĻglement proposÃĐ ÃĐlargirait la portÃĐe des conditions au-delà des exigences relatives aux essais, oÃđ des conditions peuvent Être imposÃĐes ou modifiÃĐes au moment de lâhomologation ou aprÃĻs lâhomologation et devraient se rapporter principalement aux incertitudes liÃĐes aux avantages et/ou aux risques dâun instrument mÃĐdical.
On sâattend à ce que les conditions puissent Être utilisÃĐes pour ÃĐvaluer lâinnocuitÃĐ ou lâefficacitÃĐ Ã long terme dâun instrument mÃĐdical en rÃĐponse à des preuves, y compris des preuves rÃĐelles, qui ne sont pas disponibles au moment de la dÃĐlivrance de lâhomologation. Elles pourraient ÃĐgalement Être utilisÃĐes pour recueillir des donnÃĐes spÃĐcifiques aprÃĻs lâhomologation pour des sous-groupes sous-reprÃĐsentÃĐs de donnÃĐes cliniques afin dâamÃĐliorer lâÃĐtiquetage ou les statistiques des sous-populations spÃĐcifiques (par exemple femmes enceintes, pÃĐdiatrie).
En moyenne, chaque annÃĐe, il y a environ 1 810 licences dâinstruments mÃĐdicaux homologuÃĐs et 150 conditions ÃĐmises. En vertu du rÃĻglement proposÃĐ, on prÃĐvoit une augmentation de 5 % à 10 % des conditions ÃĐmises. Par souci de prudence, on suppose que les conditions augmenteraient de 10 % ou que 15 conditions supplÃĐmentaires seraient ÃĐmises par annÃĐe. De plus, les donnÃĐes historiques ont montrÃĐ que des conditions sont habituellement rÃĐglÃĐes à lâintÃĐrieur de trois ans. Tout comme pour le traitement des conditions pour les drogues, le coÃŧt de conformitÃĐ aux conditions des instruments mÃĐdicaux serait rÃĐparti sur la pÃĐriode de trois ans.
Selon les rÃĐponses au sondage dâÃĐtablissement des coÃŧts, les coÃŧts de lâexÃĐcution des conditions varient de 4 000 $ à 1 million de dollars. Cela est basÃĐ sur lâexpÃĐrience des intervenants à lâÃĐgard des conditions y compris, sans sây limiter, leurs activitÃĐs allant de la prÃĐsentation des rÃĐsultats dâune ÃĐtude finale, des donnÃĐes non cliniques supplÃĐmentaires et/ou des rÃĐsultats dâune ÃĐtude en cours à une nouvelle ÃĐtude clinique pour confirmer lâefficacitÃĐ. Le coÃŧt moyen de lâexÃĐcution des conditions est de 218 000 $.
Les coÃŧts prÃĐvus des conditions pour les instruments mÃĐdicaux au cours de la premiÃĻre annÃĐe de mise en Åuvre (deuxiÃĻme annÃĐe du rÃĻglement) sont de 1 090 000 $, de 2 180 000 $ pour la deuxiÃĻme annÃĐe et de 3 270 000 $ pour chaque annÃĐe suivante. Cela se traduit par une VA de 18 315 378 $ sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Plans de gestion des risques
Le rÃĻglement proposÃĐ officialiserait la pratique de demander des PGR aux fabricants de drogues pour usage humain. ConformÃĐment à la pratique actuelle, les fabricants seraient tenus dâinclure des renseignements propres au Canada, selon le contexte et le marchÃĐ canadien. Le ministre pourrait ÃĐgalement exiger un PGR à jour sâil y avait des motifs raisonnables de croire que les risques et les incertitudes associÃĐs à la drogue sont considÃĐrablement diffÃĐrents du plan existant ou si la drogue prÃĐsente un risque grave de prÃĐjudice à la santÃĐ humaine qui justifie des mesures visant à rÃĐduire la possibilitÃĐ ou la gravitÃĐ dâun tel prÃĐjudice qui sont considÃĐrablement diffÃĐrentes de celles dÃĐcrites dans le plan actuel. Les modifications crÃĐeraient ÃĐgalement une nouvelle obligation pour les fabricants de soumettre un rÃĐsumÃĐ des PGR nouveaux et mis à jour dans les deux langues officielles.
à lâheure actuelle, SantÃĐ Canada reçoit en moyenne 103 nouveaux PGR par annÃĐe, conformÃĐment à la ligne directrice. Tous ces nouveaux PGR sont soumis volontairement sur demande. Sur ces 103 PGR, la plupart ont dÃĐjà fourni des rÃĐsumÃĐs, mais seulement en anglais; par consÃĐquent, on sâattend à ce que les fabricants doivent traduire ces rÃĐsumÃĐs de PGR. De plus, SantÃĐ Canada reçoit en moyenne 321 PGR mis à jour par annÃĐe. En vertu du rÃĻglement proposÃĐ, tous les 321 PGR mis à jour exigeraient un rÃĐsumÃĐ dans les deux langues.
En rÃĐponse à lâenquÊte sur lâACA, les intervenants ont fourni une vaste gamme de coÃŧts allant de lâÃĐlaboration dâun PGR de base, à la crÃĐation dâun rÃĐsumÃĐ, à la traduction. Il est à noter que la plupart des coÃŧts fournis sont fondÃĐs sur lâexpÃĐrience rÃĐelle de lâÃĐlaboration de PGR au Canada ou dans dâautres pays. Il est reconnu que la traduction dâun rÃĐsumÃĐ mis à jour exigerait beaucoup moins de temps et dâefforts que la prÃĐparation du rÃĐsumÃĐ initial, car la plupart des changements apportÃĐs à un rÃĐsumÃĐ concernent lâajout ou la suppression dâun problÃĻme dâinnocuitÃĐ ou dâune mesure de rÃĐduction des risques. Par consÃĐquent, le coÃŧt de la traduction dâun rÃĐsumÃĐ mis à jour devrait ÃĐgalement Être infÃĐrieur. Toutefois, en raison du manque de donnÃĐes et par souci de prudence, SantÃĐ Canada utilise le mÊme coÃŧt que la traduction dâun nouveau rÃĐsumÃĐ comme approximation de coÃŧt de traduction dâun rÃĐsumÃĐ mis à jour.
Il est prÃĐvu quâun total de 424 (103 + 321) rÃĐsumÃĐs seront traduits par annÃĐe à un coÃŧt de 5 400 $ par rÃĐsumÃĐ. Cela ÃĐquivaut à 2 289 600 $ par annÃĐe, sans remise. La VA est de 14 917 276 $ sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Examens en continu
Les modifications proposÃĐes permettraient des examens en continu afin de faciliter lâaccÃĻs en temps opportun aux drogues pour usage humain et vÃĐtÃĐrinaire nÃĐcessaires pour lutter contre les maladies infectieuses ÃĐmergentes au Canada, ainsi quâaux drogues pour le traitement, la prÃĐvention ou le diagnostic de maladies ou dâaffections graves ou gravement dÃĐbilitantes.
Les exigences proposÃĐes avant la soumission de la prÃĐsentation de drogue incluent un dossier de demande dâexamen en continu, qui comprendrait des renseignements dÃĐmontrant comment la drogue à laquelle la prÃĐsentation se rapporterait satisfait aux critÃĻres dâadmissibilitÃĐ. Cela est semblable au dossier dâÃĐvaluation clinique fourni en vertu de la politique sur lâÃĐvaluation prioritaire actuelle. De plus, avant de dÃĐposer la prÃĐsentation, le fabricant serait tenu de fournir au ministre un plan de prÃĐsentation, qui prÃĐciserait comment et quand le fabricant a lâintention de fournir les renseignements manquants. On sâattend à ce que les renseignements manquants soient prÃĐsentÃĐs dans deux à quatre transactions dans le dÃĐlai ÃĐnoncÃĐ dans lâavis. Les intervenants ont indiquÃĐ que le coÃŧt estimatif pour ÃĐlaborer et soumettre un plan de prÃĐsentation se situerait entre 40 000 $ et 59 000 $. Le coÃŧt est variable en raison du fait quâun plan de prÃĐsentation est une rÃĐplique dâune prÃĐsentation dâexamen en continu à lâÃĐchelle mondiale ou est prÃĐparÃĐ pour le Canada seulement. Le coÃŧt moyen dâun plan de prÃĐsentation est prÃĐsumÃĐ ÃŠtre de 49 500 $. De plus, comme lâont indiquÃĐ les intervenants, le coÃŧt associÃĐ Ã la fourniture de renseignements manquants est dâenviron 7 500 $ par transaction. On suppose quâil y aurait cinq examens en continu de drogues pour usage humain par annÃĐe et que les renseignements manquants seraient fournis en moyenne plus de trois transactions par prÃĐsentation.
Lâexamen en continu serait une option pour certaines prÃĐsentations de drogue, puisque les fabricants peuvent plutÃīt attendre et dÃĐposer une prÃĐsentation non continue ou se fonder sur des politiques dâexamen accÃĐlÃĐrÃĐ comme lâÃĐvaluation prioritaire. Ainsi, le coÃŧt total supplÃĐmentaire estimÃĐ pour utiliser lâexamen en continu plutÃīt que dâautres options dâexamen pour cinq prÃĐsentations de drogue par annÃĐe est de 360 000 $. Le coÃŧt prÃĐvu en VA est de 2 345 484 $ sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Des examens en continu existent dÃĐjà en pratique pour les mises à jour annuelles des vaccins contre la grippe saisonniÃĻre et pour les mÃĐdicaments vÃĐtÃĐrinaires pour lesquels le ministre a indiquÃĐ son intention de mener lâexamen avec un organisme de rÃĐglementation ÃĐtranger. Les modifications fourniraient un cadre rÃĐglementaire pour ces pratiques actuelles. Dans le contexte de SantÃĐ Canada, des examens conjoints ou parallÃĻles sont effectuÃĐs avec des organismes de rÃĐglementation internationaux, notamment le Centre de mÃĐdecine vÃĐtÃĐrinaire de la FDA des Ãtats-Unis, sous la supervision du Conseil de coopÃĐration en matiÃĻre de rÃĐglementation.
Il nâest pas prÃĐvu quâil y aurait des coÃŧts supplÃĐmentaires pour les examens en continu des mises à jour annuelles des vaccins contre la grippe saisonniÃĻre ou pour les mÃĐdicaments vÃĐtÃĐrinaires pour lesquels le ministre a indiquÃĐ son intention de mener lâexamen avec un organisme de rÃĐglementation ÃĐtranger puisque le rÃĻglement proposÃĐ est conçu pour codifier ce qui a ÃĐtÃĐ fait en pratique.
PrÃĐpositionnement
Il nâest pas prÃĐvu quâil y aurait des rÃĐpercussions sur lâindustrie, car les modifications proposÃĐes continueraient de limiter le prÃĐpositionnement aux drogues semblables à celles qui traitent la COVID-19, à savoir les drogues dâurgence de santÃĐ publique. La dÃĐcision du prÃĐpositionnement dâune drogue dâurgence de santÃĐ publique continuerait dâÊtre rÃĐgie par lâadministratrice en chef de la santÃĐ publique du Canada pour les conditions ÃĐnoncÃĐes dans la Liste dâaffections qui menacent la santÃĐ publique.
Assurer la qualitÃĐ des drogues pendant la fabrication
Les modifications proposÃĐes au titre 2 de la partie C ne devraient pas engager des coÃŧts ou imposer un fardeau supplÃĐmentaire à lâindustrie. Les modifications appuieraient lâintention des exigences existantes en matiÃĻre de contrÃīle de la qualitÃĐ en clarifiant les interprÃĐtations de la conformitÃĐ aux BPF qui sont ÃĐnoncÃĐes dans les documents dâorientation de SantÃĐ Canada Lignes directrices sur les Bonnes pratiques de fabrication des drogues (GUI-0001) et Lignes directrices sur le contrÃīle environnemental lors de lâentreposage et du transport des mÃĐdicaments (GUI-0069).
Modernisation des exigences relatives aux produits biologiques
Il nâest pas prÃĐvu quâil y aurait des coÃŧts supplÃĐmentaires pour lâindustrie, car la modification proposÃĐe abrogerait les dispositions dÃĐsuÃĻtes propres aux produits allant de C.04.050 Ã C.04.683, et dâautres ÃĐlÃĐments des modifications reflÃĐteraient ÃĐgalement la pratique actuelle.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue
Il nâest pas prÃĐvu quâil y aurait des coÃŧts supplÃĐmentaires pour lâindustrie, car les rÃĻglements proposÃĐs sont conformes à la pratique actuelle.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain
Il nâest pas prÃĐvu quâil y aurait des coÃŧts supplÃĐmentaires pour lâindustrie, car les fabricants ne seraient tenus de soumettre des donnÃĐes dâessais cliniques ventilÃĐes par sous-groupes de population pour appuyer lâinnocuitÃĐ et lâefficacitÃĐ de la prÃĐsentation de drogue nouvelle (ou un supplÃĐment) que si les donnÃĐes dÃĐsagrÃĐgÃĐes ont dÃĐjà ÃĐtÃĐ prÃĐsentÃĐes à la FDA des Ãtats-Unis ou à lâEMA.
Normes
Le rÃĻglement actuel exige que les ÃĐtiquettes intÃĐrieure et extÃĐrieure de certaines drogues indiquent si soit une norme rÃĐglementaire, une norme mentionnÃĐe dans une publication à lâannexe B de la Loi, ou la norme dâun fabricant ont ÃĐtÃĐ utilisÃĐes pour une drogue. Cette exigence sâapplique aux drogues rÃĐglementÃĐes uniquement en vertu du titre 1 et celles rÃĐglementÃĐes en vertu du titre 8 de la partie C qui ne sont pas ÃĐgalement rÃĐglementÃĐes en vertu des titres 3 et 4 (câest-à -dire les produits pharmaceutiques radioactifs et biologiques). SantÃĐ Canada propose de supprimer cette exigence en matiÃĻre dâÃĐtiquetage. On ne sâattend pas à ce que cet ÃĐlÃĐment de la proposition entraÃŪne des coÃŧts supplÃĐmentaires pour lâindustrie. Le retrait de cette exigence en matiÃĻre dâÃĐtiquetage permettrait aux fabricants dâavoir lâespace nÃĐcessaire pour fournir des renseignements qui sont plus pertinents pour les fournisseurs de soins de santÃĐ et les patients.
à lâheure actuelle, si la norme dâun fabricant est utilisÃĐe, le RÃĻglement exige que la drogue respecte le degrÃĐ maximal de puretÃĐ et la variation de puissance la plus petite de toute pharmacopÃĐe dans laquelle elle est inscrite à lâannexe B de la Loi. Ce rÃĻglement sâapplique aux drogues rÃĐglementÃĐes uniquement en vertu du titre 1 et celles rÃĐglementÃĐes en vertu du titre 8 (y compris celles qui sont ÃĐgalement rÃĐglementÃĐes en vertu des titres 3 et 4 [câest-à -dire les produits pharmaceutiques radioactifs et biologiques]). Il est proposÃĐ que les drogues rÃĐglementÃĐes en vertu du titre 8 (autre que celles ÃĐnumÃĐrÃĐes à lâannexe C de la Loi) à lâÃĐgard duquel la norme dâun fabricant est revendiquÃĐe seraient exemptÃĐes de ce rÃĻglement (par exemple les fabricants seraient autorisÃĐs à nuancer les limites de puretÃĐ et/ou de puissance au-delà de celles indiquÃĐes dans une pharmacopÃĐe de lâannexe B qui seraient acceptables pour SantÃĐ Canada). On ne sâattend pas à ce que cet ÃĐlÃĐment du projet entraÃŪne des coÃŧts supplÃĐmentaires pour lâindustrie. On sâattend en plus à ce que cet ÃĐlÃĐment rÃĐduise le fardeau rÃĐglementaire inutile.
CoÃŧts pour le gouvernement
Conditions pour les drogues et instruments mÃĐdicaux
Il existe une incertitude quant à la frÃĐquence et le type de conditions que SantÃĐ Canada pourrait ÃĐmettre à lâavenir. Il y a deux facteurs qui pourraient crÃĐer une augmentation du nombre de conditions ÃĐmises. La premiÃĻre est une augmentation du nombre de prÃĐsentations de drogue dans des domaines oÃđ il existe des besoins non satisfaits, comme lâoncologie ou les maladies rares pÃĐdiatriques (oÃđ la probabilitÃĐ dâappliquer des conditions est relativement ÃĐlevÃĐe). La deuxiÃĻme est une augmentation gÃĐnÃĐrale de lâutilisation des conditions une fois que les autorisations gÃĐnÃĐrales des conditions pour les drogues et les instruments mÃĐdicaux entrent en vigueur, en particulier lorsque les incertitudes ou les risques nâont peut-Être pas ÃĐtÃĐ cernÃĐs au moment de lâautorisation. LâACA constate une augmentation initiale du nombre de conditions ÃĐmises en raison du rÃĻglement proposÃĐ, mais on ne sâattend pas à ce quâil y ait une augmentation supplÃĐmentaire au cours de la pÃĐriode dâanalyse parce que de nombreux facteurs (comme la faisabilitÃĐ, des moyens moins contraignants ou lâapplication des exigences existantes) seraient pris en compte avant dâimposer des conditions.
Le coÃŧt pour le gouvernement de la gestion des conditions est trÃĻs variable et dÃĐpend du risque du produit ÃĐvaluÃĐ et des exigences stipulÃĐes dans les conditions. Le coÃŧt de la gestion de ces conditions varie de 119 994 $ à 167 982 $, soit en moyenne 143 988 $, par condition imposÃĐe au moment de la dÃĐlivrance de lâautorisation de mise en marchÃĐ. Le coÃŧt de la gestion des conditions imposÃĐes aprÃĻs lâautorisation varie de 159 568 $ à 180 562 $, soit en moyenne 170 064 $ par condition. On prÃĐvoit que 10 conditions pour les drogues (5 au moment de lâautorisation et 5 aprÃĻs lâautorisation) seraient gÃĐrÃĐes annuellement par SantÃĐ Canada, ce qui reprÃĐsente un coÃŧt annuel de 1 570 266 $ ou 10 230 649 $ VA sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
On dÃĐnombre environ 150 instruments mÃĐdicaux homologuÃĐs qui font lâobjet de conditions en vertu du RIM et on estime que cela augmenterait de 5 à 10 % par annÃĐe en raison de la portÃĐe ÃĐlargie des conditions. Si la limite supÃĐrieure ÃĐtait utilisÃĐe, environ 15 nouvelles conditions seraient ÃĐmises par annÃĐe à un coÃŧt moyen de 105 046 $, ce qui entraÃŪnerait des coÃŧts de gestion dâenviron 1 575 698 $ par annÃĐe ou 10 266 035 $ VA sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Des coÃŧts supplÃĐmentaires pour le gouvernement peuvent ÃĐgalement Être engagÃĐs lorsque des mesures de vÃĐrification de la conformitÃĐ ou dâapplication de la loi sont prises relativement aux conditions. Le choix par SantÃĐ Canada dâune mesure particuliÃĻre de conformitÃĐ et dâapplication de la loi est informÃĐ par une approche fondÃĐe sur le risque et dÃĐpendrait du type de condition. On sâattend à ce que les mesures dâapplication de la loi soient rares et rÃĐservÃĐes pour les cas oÃđ il est nÃĐcessaire de traiter une violation connue ou soupçonnÃĐe de condition.
Le coÃŧt total pour le gouvernement de la gestion des conditions pour les drogues et les instruments mÃĐdicaux devrait Être de 3 145 964 $ par annÃĐe ou de 20 496 684 $ VA sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
Plans de gestion des risques
SantÃĐ Canada reçoit actuellement des PGR volontairement, or le gouvernement nÃĐgocie souvent avec les demandeurs pour harmoniser le contenu des PGR avec ce qui est dÃĐcrit dans les lignes directrices, particuliÃĻrement en vue de lâinclusion dâun addenda portant sur des renseignements propres au Canada. à lâheure actuelle, les recommandations et les commentaires fournis par SantÃĐ Canada sont abordÃĐs environ 90 % du temps, 70 % de ces PGR ÃĐtant ÂŦ acceptÃĐs avec des changements mineurs Âŧ. Presque tous les fabricants incluent un rÃĐsumÃĐ avec leur prÃĐsentation de PGR et environ 40 % incluent un addenda de renseignements propres au Canada.
Des coÃŧts supplÃĐmentaires pour le gouvernement peuvent dÃĐcouler dâactivitÃĐs de conformitÃĐ et dâapplication de la loi qui seraient nÃĐcessaires pour remÃĐdier à lâincapacitÃĐ dâun organisme de prÃĐsenter un plan mis à jour conformÃĐment au rÃĻglement proposÃĐ, mais on sâattend à ce que ces cas soient rares.
Examens en continu
Les modifications proposÃĐes permettraient dâoffrir lâoption dâun examen en continu pour les prÃĐsentations de drogue nouvelle et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour certaines drogues. Bien que les modifications permettraient à un fabricant de fournir des composantes de la prÃĐsentation dans des transactions sÃĐquentielles à mesure que lâinformation devient disponible aprÃĻs le dÃĐpÃīt de la prÃĐsentation de drogue, la norme et le seuil de donnÃĐes requis pour obtenir une autorisation seraient les mÊmes que pour une prÃĐsentation examinÃĐe sans examen en continu. MÊme si lâinformation requise pour un examen en continu ÃĐtait exactement la mÊme que pour une prÃĐsentation de drogue nouvelle non continue ou un supplÃĐment à une prÃĐsentation de drogue nouvelle non continue, le processus dâexamen devrait exiger davantage de ressources pour SantÃĐ Canada en raison de lâapproche progressive de lâexamen.
Chaque nouvelle transaction reçue conformÃĐment à lâoption dâexamen en continu devrait exiger un dÃĐlai supplÃĐmentaire pour :
- les rÃĐvisions des PGR (une fois adoptÃĐes dans les rÃĻglements);
- les interactions accrues pendant lâÃĐtape prÃĐalable à la mise en marchÃĐ;
- les communications des promoteurs (câest-à -dire que lâexamen de la prÃĐsentation reprendrait à des moments diffÃĐrents à mesure que lâinformation est fournie aux fins dâexamen);
- lâexamen du matÃĐriel ÃĐducatif;
- les commentaires additionnels sur lâÃĐtiquetage;
- le dÃĐlai dâexamen supplÃĐmentaire pour approbation à lâÃĐtape de lâexamen final.
De nouveaux coÃŧts ponctuels dÃĐcouleraient ÃĐgalement des rÃĐvisions nÃĐcessaires aux documents de procÃĐdures opÃĐrationnelles normalisÃĐes (PON) pour inclure les mises à jour de la formation, lâÃĐlaboration de processus et de matÃĐriels de processus et une nouvelle surveillance. Ces coÃŧts sont estimÃĐs à 131 888 $ au cours de la premiÃĻre annÃĐe.
Tous les coÃŧts supplÃĐmentaires sont supposÃĐs Être attribuables à un examen non contigu des renseignements fournis à lâappui de la prÃĐsentation, ainsi quâau traitement des renseignements manquants aprÃĻs le dÃĐpÃīt de la prÃĐsentation.
Bien que le contenu dâune prÃĐsentation aux fins dâexamen en continu soit le mÊme que pour la prÃĐsentation ÃĐquivalente sans examen en continu, en raison de la nature pÃĐriodique de lâexamen et de lâuniformitÃĐ requise dans lâensemble du processus dâexamen (de prÃĐfÃĐrence par la mÊme ÃĐquipe dâexamen), le coÃŧt supplÃĐmentaire pour chaque prÃĐsentation est estimÃĐ Ã 127 179 $ pour SantÃĐ Canada. En supposant quâil y ait 5 prÃĐsentations, le coÃŧt total prÃĐvu au cours des annÃĐes subsÃĐquentes est de 635 895 $ ou 4 274 892 $ VA sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
De plus, il y a un coÃŧt ponctuel prÃĐvu de 1 028 207 $ pour tenir compte dâune mise à jour de lâinfrastructure des technologies de lâinformation (TI). Cette mise à jour serait nÃĐcessaire pour traiter les transactions supplÃĐmentaires pour les examens en continu, les conditions et les PGR. Ce coÃŧt devrait ÃĐgalement tenir compte des mises à jour dans docuBridge (un systÃĻme de gestion des prÃĐsentations), des changements apportÃĐs aux formulaires de demande, de la formation et de la mise en Åuvre. Pour chaque annÃĐe subsÃĐquente, il y aurait ÃĐgalement un coÃŧt de 65 887 $ pour gÃĐrer les transactions additionnelles pour les examens en continu, les conditions et les PGR. Cela se traduit à un coÃŧt total de 1 457 476 $ VA pour les solutions de TI sur une pÃĐriode de 10 ans, actualisÃĐe à 7 %.
PrÃĐpositionnement
Les modifications proposÃĐes permettraient quâune drogue soit prÃĐpositionnÃĐe non seulement en rÃĐponse à la pandÃĐmie de COVID-19, mais pour dâautres urgences de santÃĐ publique qui seraient mentionnÃĐes dans la Liste dâaffections qui menacent la santÃĐ publique. Les modifications proposÃĐes ne devraient pas donner lieu à la prÃĐsentation ou à lâexamen de renseignements nouveaux ou supplÃĐmentaires et nâauraient pas dâincidence sur les activitÃĐs actuelles du gouvernement. Les coÃŧts prÃĐvus devraient Être nÃĐgligeables.
Assurer la qualitÃĐ des drogues pendant la fabrication
Aucun changement nâest prÃĐvu à la surveillance des drogues par SantÃĐ Canada ou à la pratique opÃĐrationnelle actuelle en ce qui concerne les inspections des BPF à la suite des modifications proposÃĐes. Aucun coÃŧt supplÃĐmentaire nâest prÃĐvu pour le gouvernement.
Modernisation des exigences relatives aux produits biologiques
Les modifications visent à intÃĐgrer les pratiques opÃĐrationnelles et industrielles dans les rÃĻglements. Ãtant donnÃĐ que la rÃĐfÃĐrence de lâanalyse des coÃŧts est une pratique actuelle, on ne prÃĐvoit aucun coÃŧt supplÃĐmentaire pour le gouvernement à la suite de ces modifications.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue
Aucun coÃŧt pour le gouvernement nâa ÃĐtÃĐ identifiÃĐ.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain
On ne prÃĐvoit pas quâil y aurait une incidence sur le nombre de prÃĐsentations à la suite de cette modification rÃĐglementaire. Lâexigence proposÃĐe pourrait amÃĐliorer la qualitÃĐ des donnÃĐes soumises, car les examinateurs analysent dÃĐjà les donnÃĐes dÃĐsagrÃĐgÃĐes fournies volontairement, et ces donnÃĐes continueraient dâÊtre examinÃĐes comme câest le cas actuellement. Il pourrait y avoir une autre ÃĐtape ajoutÃĐe à lâexamen de la prÃĐsentation, mais le travail supplÃĐmentaire ne devrait pas Être important et serait gÃĐrÃĐ par le personnel existant. Une formation des examinateurs serait requise ainsi que lâÃĐlaboration des PON, mais les ressources requises font partie de la mise en Åuvre du plan dâaction sur lâanalyse comparative fondÃĐe sur le sexe et le genre plus de la Direction gÃĐnÃĐrale des produits de santÃĐ et des aliments.
Normes
Aucun coÃŧt pour le gouvernement nâa ÃĐtÃĐ identifiÃĐ.
Avantages
Les modifications proposÃĐes moderniseraient le libellÃĐ dans le RAD et le RIM, apporteraient une clartÃĐ Ã lâindustrie et à lâorganisme de rÃĐglementation, ce qui permettra une plus grande souplesse pour faire face aux innovations futures. Bien quâil soit reconnu que bon nombre des dispositions proposÃĐes dans ces modifications ne font que codifier la pratique existante, la clartÃĐ fournie devrait favoriser une plus grande innovation et un accÃĻs plus rapide aux produits au Canada et fournir des gains dâefficacitÃĐ Ã lâindustrie, aux payeurs et aux professionnels de la santÃĐ tout en amÃĐliorant lâinnocuitÃĐ et lâefficacitÃĐ, et en permettant de dissiper lâincertitude.
CombinÃĐs, ces rÃĻglements proposÃĐs contribueraient à lâapproche du cycle de vie de la rÃĐglementation des produits thÃĐrapeutiques nÃĐcessaire pour aider SantÃĐ Canada à trouver un ÃĐquilibre entre la protection des Canadiens contre les effets nocifs des produits thÃĐrapeutiques quâils utilisent tout en leur offrant des avantages thÃĐrapeutiques.
Avantages pour lâindustrie
Conditions pour les drogues
Le coÃŧt de mise au point dâune drogue sur le marchÃĐ est estimÃĐ entre 800 millions de dollars et 2,5 milliards de dollars lorsque tous les coÃŧts de recherche et de dÃĐveloppement, y compris les ÃĐchecs des drogues, sont inclus. Le processus prend entre 9 et 15 ans de la dÃĐcouverte à lâapprobationrÃĐfÃĐrence 3. Au cours du processus de mise au point des drogues, environ 60 % de toutes les drogues ÃĐchouent en raison du manque de preuves dâinnocuitÃĐ et dâefficacitÃĐ. Une fois la drogue sur le marchÃĐ, son rendement dans des conditions rÃĐelles peut Être diffÃĐrent de celui dâun milieu clinique. Un problÃĻme important dâinnocuitÃĐ ou dâefficacitÃĐ met non seulement la santÃĐ et la sÃĐcuritÃĐ des patients en danger, mais le retrait subsÃĐquent dâune drogue du marchÃĐ pourrait coÃŧter des millions de dollars à lâindustrie. Dans certains cas, la drogue pourrait avoir ÃĐtÃĐ trÃĻs bÃĐnÃĐfique à un groupe de la population tout en posant un risque pour un autre groupe. Les modifications rÃĐglementaires proposÃĐes visent à prÃĐciser les dispositions existantes, à offrir un accÃĻs plus rapide ou plus flexible au marchÃĐ, à prÃĐvenir les pÃĐnuries et à aider à gÃĐrer les incertitudes et les risques afin de rÃĐduire la probabilitÃĐ de dÃĐfaillance du produit.
Lâapplication des conditions imposÃĐes à une identification numÃĐrique dâune drogue, avant ou aprÃĻs lâautorisation, en plus de lâutilisation des PGR, aiderait à cerner plus rapidement les problÃĻmes dâinnocuitÃĐ et dâefficacitÃĐ, permettant ainsi à SantÃĐ Canada dâappliquer les stratÃĐgies dâattÃĐnuation des risques pour rÃĐpondre aux signaux de sÃĐcuritÃĐ provenant de conditions rÃĐelles afin de rÃĐduire les dommages, tout en permettant le maintien dâun produit sur le marchÃĐ avec des contrÃīles de sÃĐcuritÃĐ appropriÃĐs, au besoin. Ceci permettrait à lâindustrie de maintenir un produit sur le marchÃĐ sans interruption.
Conditions pour les instruments mÃĐdicaux
Les modifications renforceraient la capacitÃĐ de SantÃĐ Canada à assurer une surveillance, une ÃĐvaluation et une communication continues que ce soit à lâÃĐtape de lâhomologation ou aprÃĻs la mise en marchÃĐ des instruments mÃĐdicaux. Pour lâindustrie, cela pourrait signifier lâidentification ainsi que la rÃĐsolution et lâattÃĐnuation rapides des risques qui surviennent tout en permettant aux appareils de rester sur le marchÃĐ sans interruption.
Plans de gestion des risques
Aucun avantage pour lâindustrie nâa ÃĐtÃĐ identifiÃĐ.
Examens en continu
Pour lâexamen en continu des drogues comme les vaccins contre la grippe saisonniÃĻre, les modifications proposÃĐes servent à codifier la pratique existante et ne reprÃĐsentent aucun avantage, puisque lâindustrie fonctionne dÃĐjà comme si les dispositions ÃĐtaient en place.
En cas dâun ÃĐvÃĐnement majeur semblable à la pandÃĐmie de COVID-19, le RÃĻglement fournirait à lâindustrie des exigences claires pour un examen en continu afin de traiter une urgence de santÃĐ publique ajoutÃĐe à une liste, qui serait incorporÃĐe par renvoi.
Pour les drogues qui satisfont aux exigences dâadmissibilitÃĐ pour avoir accÃĻs à lâoption dâexamen en continu, un accÃĻs plus rapide au marchÃĐ pouvant aller jusquâà six mois pourrait Être possible comparativement au processus normal. à partir dâun examen des dÃĐlais dâexamen prioritaires de SantÃĐ Canada et des types de drogues qui devraient Être admissibles à un examen en continu, on estime que ces drogues seraient approuvÃĐes en moyenne deux mois plus tÃīt que dans le cadre des processus actuels. Bien quâil puisse y avoir des coÃŧts associÃĐs au fait dâÊtre sur le marchÃĐ pendant deux mois supplÃĐmentaires, la majoritÃĐ des coÃŧts impliquÃĐs sont censÃĐs Être supportÃĐs pour se rendre sur le marchÃĐ; par consÃĐquent, la valeur des ventes est utilisÃĐe comme mesure de lâavantage. En 2019, avant la pandÃĐmie, les ventes des 1 364 drogues brevetÃĐes au Canada ont atteint 17,2 milliards de dollars, ce qui donne un revenu annuel moyen de 12,6 millions de dollars par drogue brevetÃĐerÃĐfÃĐrence 4. La moyenne des 10 principales ventes de drogues au Canada en 2020 dÃĐpassait 500 millions de dollars, les ventes les plus ÃĐlevÃĐes dÃĐpassant un milliard de dollarsrÃĐfÃĐrence 5.
En supposant que cinq drogues, dont les ventes annuelles moyennes (12 600 000 $) ont ÃĐtÃĐ approuvÃĐes deux mois plus tÃīt (17 %), lâoption dâexamen en continu pourrait offrir un avantage à lâindustrie de 10,7 millions de dollars en ventes.
5 drogues à 12 600 000 $ à 17 % = 10 710 000 $
Lâavantage quantifiable prÃĐvu pour lâindustrie est de 10 710 000 $ annuellement ou de 69 788 137 $ VA sur une pÃĐriode de 10 ans.
Assurer la qualitÃĐ des drogues pendant la fabrication
Lâavantage attendu est la clartÃĐ du RÃĻglement.
Modernisation des exigences relatives aux produits biologiques
Ãtant donnÃĐ que les modifications rÃĐglementaires servent à moderniser le RÃĻglement et quâelles reflÃĐteraient la pratique actuelle, lâavantage prÃĐvu est une plus grande clartÃĐ pour lâindustrie.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue
Lâavantage attendu est la clartÃĐ de lâintention rÃĐglementaire.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain
Aucun avantage pour lâindustrie nâa ÃĐtÃĐ identifiÃĐ.
Normes
Les fabricants nâauraient plus à modifier leur ÃĐtiquette sâil devenait nÃĐcessaire pour eux de changer la norme de leur drogue. Le projet libÃĐrerait de lâespace sur lâÃĐtiquette, ce qui donnerait aux fabricants la souplesse nÃĐcessaire pour fournir dâautres renseignements utiles aux patients et aux professionnels de la santÃĐ. En outre, pour les drogues qui revendiquent la norme dâun fabricant, les modifications pourraient rÃĐduire le temps que les fabricants doivent consacrer à la surveillance de la pharmacopÃĐe et le coÃŧt des nouveaux essais de drogues pour respecter les normes les plus strictes.
Bien que lâavantage ne se limite pas uniquement aux fabricants de mÃĐdicaments gÃĐnÃĐriques, ils seraient plus susceptibles de bÃĐnÃĐficier de la modification rÃĐglementaire proposÃĐe. Les intervenants ont indiquÃĐ des ÃĐconomies allant de 120 000 $ à 400 000 $ par annÃĐe; cela inclurait les coÃŧts dâÃĐlimination des ÃĐtiquettes. De plus, les ÃĐconomies rÃĐalisÃĐes grÃĒce aux normes du fabricant pourraient varier de 45 000 $ à 650 000 $ par annÃĐe en ce qui a trait à la surveillance et aux nouveaux essais de drogues. Les ÃĐconomies de coÃŧts administratifs liÃĐes à la surveillance des pharmacopÃĐes de lâannexe B, à lâÃĐvaluation des impacts et à la dÃĐclaration à SantÃĐ Canada sont estimÃĐes à environ 37,5 heures pour chaque pharmacopÃĐe et supposent quâil y a trois publications par annÃĐe. En adoptant une approche moyenne, les ÃĐconomies annuelles rÃĐalisÃĐes grÃĒce à lâÃĐtiquetage et aux normes du fabricant sont dâenviron 260 000 $ et de 347 500 $ respectivement. Ãtant donnÃĐ quâil y a 15 entreprises qui fournissent des produits gÃĐnÃĐriques au Canada, on estime que les ÃĐconomies dÃĐcoulant du rÃĻglement proposÃĐ pourraient sâÃĐlever à 9 112 500 $ par annÃĐe.
15 entreprises à (260 000 $ + 347 500 $) = 9 112 500 $
On sâattend à ce que la modification proposÃĐe permette de rÃĐaliser des ÃĐconomies annuelles de 9 112 500 $ ou de 68 482 554 $ VA sur une pÃĐriode de 10 ans. De plus, les intervenants ont ÃĐgalement indiquÃĐ que le rÃĻglement actuel cause parfois des pÃĐnuries, puisque les nouvelles normes doivent Être respectÃĐes. Le rÃĻglement proposÃĐ rÃĐduirait ces perturbations des ventes.
Avantages pour le gouvernement
Conditions pour les drogues
Les modifications codifieraient dans la loi les pratiques existantes en vertu de la politique sur les AC-C et permettraient lâimposition de conditions au moment de la dÃĐlivrance de lâautorisation de mise en marchÃĐ et aprÃĻs lâautorisation. Elles pourraient rÃĐduire le temps nÃĐcessaire pour parvenir à une solution dans les discussions avec un fabricant sur la façon de traiter avec les problÃĻmes potentiels dâinnocuitÃĐ et dâincertitude dâun produit. Les modifications pourraient ÃĐgalement permettre à SantÃĐ Canada de sâadapter à lâinnovation et au changement.
Conditions pour les instruments mÃĐdicaux
Les modifications codifieraient dans la loi les pratiques existantes en matiÃĻre de conditions à SantÃĐ Canada et ÃĐlargiraient la portÃĐe des conditions pouvant Être imposÃĐes sur une homologation dâinstrument mÃĐdical. Les modifications pourraient ÃĐgalement permettre à SantÃĐ Canada de sâadapter à lâinnovation et au changement.
Plans de gestion des risques
SantÃĐ Canada serait capable dâobliger les fabricants à soumettre et à modifier des PGR pour combler toute lacune en temps opportun lorsquâils ne lâauraient pas fait volontairement.
Examens en continu
Au cours des 20 derniÃĻres annÃĐes, le Canada a connu trois perturbations majeures sous la forme de maladies infectieuses : le SRAS en 2003, la H1N1 en 2009 et la COVID-19 en 2020. Les drogues utilisÃĐes pour traiter ces types dâÃĐclosions de maladies transmissibles pourraient Être approuvÃĐes au moyen de lâoption dâexamen en continurÃĐfÃĐrence 6,rÃĐfÃĐrence 7,rÃĐfÃĐrence 8. Les modifications au rÃĻglement permettraient probablement lâutilisation de lâoption dâexamen en continu pour les drogues servant à combattre ces types de pandÃĐmies, sans avoir recours à des arrÊtÃĐs dâurgence.
Lâutilisation dâexamens en continu pour les vaccins contre la grippe saisonniÃĻre est une pratique courante bien ÃĐtablie et aucun avantage supplÃĐmentaire nâest identifiÃĐ.
Modernisation des exigences relatives aux produits biologiques
Des rÃĻglements modernes et clairement rÃĐdigÃĐs peuvent rÃĐduire le nombre de demandes de renseignements concernant les dispositions, ce qui permettrait à SantÃĐ Canada de gagner du temps et des ressources.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue
Ces modifications proposÃĐes clarifieraient le pouvoir du ministre de tenir compte, dans le cadre dâune prÃĐsentation, des renseignements obtenus directement ou indirectement dâun bÃĒtiment ou dâun site oÃđ une drogue est fabriquÃĐe, emballÃĐe, ÃĐtiquetÃĐe ou analysÃĐe, lorsquâil prend la dÃĐcision dâautoriser ou non la drogue.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain
Lâexigence proposÃĐe pourrait accroÃŪtre lâutilitÃĐ des donnÃĐes soumises, en rendant plus ÃĐvidente lâidentification de toute diffÃĐrence potentielle dâinnocuitÃĐ ou dâefficacitÃĐ entre les sous-populations.
Normes
Aucun avantage pour le gouvernement nâa ÃĐtÃĐ cernÃĐ.
Avantages aux Canadiens
Avantages combinÃĐs
Lâutilisation de conditions pour les drogues et les instruments mÃĐdicaux et de PGR pour les drogues, ainsi que la rÃĐception de donnÃĐes dÃĐsagrÃĐgÃĐes, augmenteraient lâinnocuitÃĐ de ces produits thÃĐrapeutiques au Canada, et pourraient aider à rÃĐduire lâincidence des rÃĐactions indÃĐsirables à un mÃĐdicament (RIM) et des incidents liÃĐs aux matÃĐriels mÃĐdicaux (IMM), et à gÃĐrer les incertitudes quant à lâinnocuitÃĐ et à lâefficacitÃĐ des mÃĐdicaments et des instruments dans des conditions rÃĐelles. Si ces mesures ÃĐtaient utilisÃĐes conjointement avec un examen en continu, il serait possible que de nouvelles drogues dont on a besoin de toute urgence obtiennent une autorisation de mise sur le marchÃĐ plus tÃīt.
Conditions pour les drogues
La possibilitÃĐ dâimposer des conditions au moment de lâautorisation est actuellement limitÃĐe aux drogues dÃĐsignÃĐes contre la COVID-19 et à certains opioÃŊdes. En vertu du rÃĻglement proposÃĐ, lâutilisation ÃĐlargie des conditions pour toutes les drogues au moment de lâautorisation, ainsi que durant la phase suivant lâautorisation, devrait amÃĐliorer lâinnocuitÃĐ des produits.
MalgrÃĐ des ÃĐvaluations rigoureuses, la surveillance de lâinnocuitÃĐ et de lâefficacitÃĐ dans des conditions rÃĐelles permet souvent dâidentifier les prÃĐjudices et les incertitudes qui nâont pas ÃĐtÃĐ dÃĐmontrÃĐs lors des ÃĐvaluations cliniques prÃĐalables à la mise en marchÃĐ. En 2019, SantÃĐ Canada a reçu plus de 96 000 rapports de RIM à lâÃĐchelle nationalerÃĐfÃĐrence 9 liÃĐs aux mÃĐdicaments et on estime que seulement 10 % de toutes les RIM sont signalÃĐsrÃĐfÃĐrence 10. Selon les estimations, jusquâà 1 Canadien sur 1 000 de moins de 65 ans ira à lâhÃīpital en raison dâune RIM au cours de leur vie. Le nombre passe à 1 sur 200 pour les Canadiens de plus de 65 ansrÃĐfÃĐrence 11. Sur plus de 96 000 signalÃĐs à SantÃĐ Canada, 18 852 signalements ont mentionnÃĐ une hospitalisation et peuvent contribuer jusquâà 6 119 dÃĐcÃĻsrÃĐfÃĐrence 9. Les femmes sont ÃĐgalement plus susceptibles que les hommes de subir une RIMrÃĐfÃĐrence 12. Les mÃĐdicaments pÃĐdiatriques ont rarement les mÊmes informations que les mÃĐdicaments pour adultes, compte tenu des limites ÃĐthiques ÃĐvidentes des tests de produits sur les enfants. Dâautres populations qui sont gÃĐnÃĐralement sous-reprÃĐsentÃĐes dans les essais cliniques menÃĐs dans le cadre de la prÃĐparation dâune prÃĐsentation de drogue rÃĐglementaire sont ÃĐgalement susceptibles de voir des avantages.
Le coÃŧt mÃĐdical direct des RIM a ÃĐtÃĐ calculÃĐ comme ÃĐtant aussi ÃĐlevÃĐ que 20 milliards de dollars (dollars de 2019) par annÃĐerÃĐfÃĐrence 13. DâaprÃĻs les donnÃĐes dÃĐclarÃĐes à SantÃĐ Canada en 2019, 19,5 % des RIM ÃĐtaient liÃĐs à lâhospitalisation et 2,6 % dÃĐclaraient une affection potentiellement mortellerÃĐfÃĐrence 14. Bien quâil ne soit pas possible de dÃĐterminer dans quelle mesure les modifications rÃĐglementaires proposÃĐes rÃĐduiront la frÃĐquence des RIM au Canada, lâincidence potentielle pourrait Être importante. Par exemple, mÊme une diminution de 0,1 % des RIM rÃĐduirait le nombre de dÃĐcÃĻs de 6 (6 119 à 0,1 %), ce qui, lorsquâon lâapplique à la valeur de 8,59 millions de dollars dâune vie statistique utilisÃĐe au CanadarÃĐfÃĐrence 15, reprÃĐsenterait un avantage de 50 millions de dollars. De mÊme, une diminution de 0,1 % du coÃŧt mÃĐdical annuel direct estimÃĐ Ã 20 milliards de dollars reprÃĐsenterait une ÃĐconomie de 20 millions de dollars par annÃĐe. Toutefois, il nâest pas possible dâestimer la rÃĐduction des RIM qui pourrait rÃĐsulter de ce rÃĻglement.
En plus du coÃŧt direct dâune RIM pour le systÃĻme de soins de santÃĐ ou pour une personne, les membres de la famille et les autres aidants familiaux doivent prendre un congÃĐ pour prendre soin dâune personne ÃĒgÃĐe, dâun enfant ou dâautres personnes qui ont ÃĐtÃĐ hospitalisÃĐes ou qui sont atteintes dâune dÃĐficience. Une rÃĐduction du nombre de RIM rÃĐsultant dâune meilleure surveillance du cycle de vie rÃĐduirait les difficultÃĐs liÃĐes à la nÃĐcessitÃĐ de prendre soin dâune personne. Les RIM provoquent ÃĐgalement de lâabsentÃĐisme et du prÃĐsentÃĐisme (travailler lorsquâon est malade avec une productivitÃĐ rÃĐduite) au travail, ce qui a une incidence sur lâactivitÃĐ ÃĐconomique.
Conditions pour les instruments mÃĐdicaux
LâÃĐlargissement des conditions permettrait de rÃĐgler les incertitudes et les risques qui ne deviennent apparents que si un instrument mÃĐdical est utilisÃĐ dans de conditions rÃĐelles. Cela pourrait Être utile pour identifier les populations de patients qui bÃĐnÃĐficieraient le plus de lâattÃĐnuation des risques graves pour la santÃĐ. Lâimposition de conditions pourrait rÃĐduire les IMM et pourrait empÊcher le retrait dâun produit du marchÃĐ qui aurait pu Être utilisÃĐ de façon sÃĐcuritaire avec les stratÃĐgies dâattÃĐnuation des risques appropriÃĐes.
Plans de gestion des risques
Aucun avantage pour les Canadiens nâa ÃĐtÃĐ identifiÃĐ.
Examens en continu
On sâattend à ce que les Canadiens bÃĐnÃĐficient dâexamens en continu pour lâapprobation de drogues pour usage humain et vÃĐtÃĐrinaire admissibles. Dans le cadre du statu quo, tous les renseignements requis pour que le ministre puisse ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ dâune nouvelle drogue sont disponibles pour un examen au moment de la prÃĐsentation. La modification rÃĐglementaire pourrait permettre à des drogues et à des vaccins spÃĐcifiques dâarriver à la conclusion du processus dâexamen de SantÃĐ Canada jusquâà six mois plus tÃīt, car lâexamen commencera avant que lâensemble complet des donnÃĐes soit disponible (la durÃĐe dÃĐpend du fait quâil soit mesurÃĐ en fonction dâun examen standard ou dâun examen prioritaire). Cela permettra une autorisation plus prÃĐcoce, ce qui pourrait mener à un accÃĻs plus rapide. Cela a le potentiel dâapporter des avantages importants en rÃĐduisant les coÃŧts des soins de santÃĐ et en amÃĐliorant la qualitÃĐ de vie dans lâensemble de la population.
SantÃĐ Canada, dâaprÃĻs son expÃĐrience de lâexamen prioritaire, estime que la crÃĐation dâune nouvelle mÃĐthode dâexamen en continu mÃĻnera à lâoctroi dâune autorisation de mise en marchÃĐ de cinq mÃĐdicaments par annÃĐe deux mois plus tÃīt. Selon le nombre de patients et le type de maladie, les avantages varieront considÃĐrablement. Pour ÃĐvaluer les avantages dâun accÃĻs plus rapide, il faut formuler des hypothÃĻses sur la taille du bassin de patients prescrits, et sur lâincidence que la drogue prescrite aura sur la qualitÃĐ de vie de ceux qui le prennent. Par souci de prudence dans son estimation, SantÃĐ Canada suppose que les drogues ciblent de petites populations de patients, comme celles qui se trouvent dans les mÃĐdicaments pour les maladies rares. Les populations de patients pourraient en fait Être beaucoup plus nombreuses dans le cas de drogues oncologiques ou de vaccins utilisant lâoption dâexamen en continu.
Une maladie rare est dÃĐfinie comme une maladie qui ne touche pas plus de 5 Canadiens sur 10 000, ou un maximum de 17 750 patients. On estime que 1 Canadien sur 12 (2,7 millions) est touchÃĐ par une maladie rare et quâil y a plus de 7 000 maladies rares dans le monde. Cela crÃĐe une taille moyenne de population de patients de 385 personnes, ce qui est, selon nous, la taille moyenne minimale de population pour toutes les drogues approuvÃĐes par cette option. Les drogues en gÃĐnÃĐral ne sont efficaces que dans 60 % de la population; câest pourquoi le nombre de patients bÃĐnÃĐficiant dâune drogue peut Être rÃĐduit à 231. Ãtant donnÃĐ que lâoption dâexamen est destinÃĐe à tous les types de drogues qui satisfont aux exigences, on suppose que cela crÃĐe un seuil infÃĐrieur pour le nombre de patients traitÃĐs.
2 700 000 personnes ÷ 7 000 maladies rares = 385 patients à 60 % = 231 patients traitÃĐs
La population du Canada entre 2015 et 2020 avait un taux de croissance moyen de 1 %. Par consÃĐquent, le nombre de patients traitÃĐs devrait ÃĐgalement augmenter de 1 % par annÃĐe. Donc, dâici la 10e annÃĐe dâanalyse, la taille des patients traitÃĐs devrait atteindre 250.
Au Canada, la valeur dâune vie statistique (VVS) utilisÃĐe dans lâanalyse ÃĐconomique est actuellement fixÃĐe par le Conseil du TrÃĐsor à 6,4 millions de dollars en dollars de 2007. En tenant compte de lâinflation, la VVS en dollars de 2021 serait de 8,59 millions de dollars. En utilisant lâespÃĐrance de vie moyenne au Canada (82 ans)rÃĐfÃĐrence 16, lâÃĒge moyen dâun Canadien (41 ans)rÃĐfÃĐrence 17, et un taux dâactualisation de 7 %, cela donne une valeur dâune annÃĐe de vie statistique (VAVS) de 599 370 $ en dollars de 2021 pour le Canadien moyenrÃĐfÃĐrence 18 en utilisant la formule suivante :
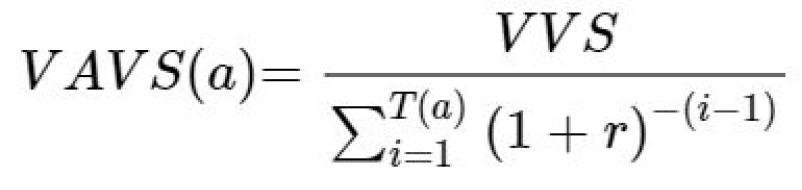
Figure Formule pour calculer la valeur de l'annÃĐe de vie statistique - Version textuelle
La VAVS Ã a est ÃĐgale au quotient de la VVS sur la somme de, parenthÃĻse ouvrante 1 plus r parenthÃĻse fermante, exposant nÃĐgatif parenthÃĻse ouvrante i moins 1 parenthÃĻse fermante, indexÃĐe de 1 Ã T parenthÃĻse ouvrante a parenthÃĻse fermante.
oÃđ VAVS est la valeur dâune annÃĐe de vie, VVS est la valeur dâune vie statistique, r est le taux dâactualisation, a est lâÃĒge et i est le nombre dâannÃĐes restantes selon lâespÃĐrance de vie à partir de lâÃĒge a.
On suppose que le rÃĻglement proposÃĐ permettra un accÃĻs rapide dâau moins 2 mois (0,17 annÃĐe). Bien que cela puisse sauver des vies dans certains cas (comme la vaccination contre la COVID-19), il est plus probable que cet accÃĻs amÃĐliorera la qualitÃĐ de vie des patients qui reçoivent un traitement. LâannÃĐe de vie ajustÃĐe en fonction de la qualitÃĐ (AVAQ) est un concept utilisÃĐ en ÃĐconomie de la santÃĐ pour tenir compte du fait que toutes les annÃĐes de vie vÃĐcues peuvent ne pas Être ÃĐgales en raison de lâexpÃĐrience de la maladie tout au long de la durÃĐe de vie. LâAVAQ de la pleine santÃĐ est dÃĐfinie comme ÃĐtant ÃĐgale à 1, et la mort comme ÃĐtant ÃĐgale à 0. Les valeurs pour dâautres ÃĐtats de maladie ont ÃĐtÃĐ calculÃĐes entre ces deux extrÃĐmitÃĐs. à tout moment de la vie, la valeur dâune annÃĐe de vie pour un individu en bonne santÃĐ est ÃĐgale à la VAVS à ce moment-là . Diverses ÃĐtudes mesurant lâAVAQ des patients souffrant de diverses maladies et affections ont gÃĐnÃĐrÃĐ un large ÃĐventail de donnÃĐes. Par exemple, lâAVAQ pour le VIH multirÃĐsistant a ÃĐtÃĐ calculÃĐe comme ÃĐtant 59 % de celle dâune personne en santÃĐ. Au moyen dâune enquÊte, les patients atteints de maladies rares traitables et leurs aidants familiaux ont dÃĐclarÃĐ avoir une AVAQ de 56 % aux Ãtats-Unis et de 50 % au Royaume-Uni, alors que leur estimation ÃĐtait de 42 % aux Ãtats-Unis et de 40 % au Royaume-Uni si la maladie ÃĐtait non traitablerÃĐfÃĐrence 19. Une rÃĐcente ÃĐtude a examinÃĐ 494 nouvelles entitÃĐs molÃĐculaires approuvÃĐes par la FDA entre 1999 et 2015 et a constatÃĐ que lâaugmentation moyenne de lâAVAQ des patients ÃĐtait de 0,38rÃĐfÃĐrence 20. Une amÃĐlioration de 0,38 de lâAVAQ pour un Canadien dâÃĒge moyen censÃĐ vivre jusquâà 82 ans reprÃĐsenterait un gain de VAVS de 227 760 $.
599 370 $ VAVS Ã 0,38 AVAQ = 227 760 $ VAVSadj
Par consÃĐquent, en supposant quâen moyenne 5 mÃĐdicaments sont approuvÃĐs chaque annÃĐe, traitant avec succÃĻs 231 patients au cours de la premiÃĻre annÃĐe pendant 2 mois supplÃĐmentaires (0,17 an) avec une AVAQ augmentÃĐe à 0,38, lâavantage de la premiÃĻre annÃĐe serait dâenviron 44,7 millions de dollars. En supposant une croissance dÃĐmographique, la valeur atteindrait 48,4 millions de dollars dâici lâannÃĐe 10.
AnnÃĐe 2 : 5 drogues à 231 patients à 0,17 annÃĐe à 227 760 $ VAVSadj = 44 720 676 $
AnnÃĐe 10 : 5 drogues à 250 patients à 0,17 annÃĐe à 227 760 $ VAVSadj = 48 426 084 $
La VA totale de lâavantage sur 10 ans serait de 301 944 819 $.
Ce nombre ne comprend pas les ÃĐconomies financiÃĻres pour les familles et les autres aidants qui consacrent du temps et des ressources à soigner les Canadiens atteints de maladies en leur apportant le soutien à la vie quotidienne (par exemple soins personnels, prÃĐparation de repas, nettoyage, transport, etc.) dont ils ont besoin.
PrÃĐpositionnement
On sâattend à ce que les Canadiens bÃĐnÃĐficient des modifications proposÃĐes. Dans le cas dâune urgence future en santÃĐ publique, le prÃĐpositionnement devrait offrir un accÃĻs rapide à des drogues propres à lâurgence. En modifiant le rÃĻglement afin dâexiger quâun importateur autorisÃĐ soit identifiÃĐ pour importer la drogue, les risques pour la santÃĐ et lâinnocuitÃĐ seraient attÃĐnuÃĐs par lâapplication dâexigences rÃĐglementaires appropriÃĐes pour appuyer la surveillance de lâactivitÃĐ dâimportation.
Modernisation des exigences relatives aux produits biologiques
Aucun avantage aux Canadiens nâest attendu, car lâinnovation se poursuivra comme elle existe actuellement selon lâorientation et la pratique actuelle.
Renseignements à lâappui de lâexamen des prÃĐsentations de drogue
Aucun avantage aux Canadiens nâest attendu, car la modification proposÃĐe reflÃĻte la pratique actuelle.
DonnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogue nouvelle pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain
Bien quâaucun avantage direct pour les Canadiens ne soit attendu en raison du projet de rÃĻglement, lâexigence de fournir des donnÃĐes dÃĐsagrÃĐgÃĐes pourrait fournir des informations supplÃĐmentaires concernant des sous-populations spÃĐcifiques qui pourraient mener à une innocuitÃĐ accrue des drogues.
Normes
Lâindustrie a indiquÃĐ que lâexigence actuelle entraÃŪne à lâoccasion des pÃĐnuries de mÃĐdicaments. Les Canadiens bÃĐnÃĐficieraient dâun approvisionnement en drogues plus stable.
Lentille des petites entreprises
La lentille des petites entreprises sâappliquerait, car tous les intervenants des industries pharmaceutiques, des instruments mÃĐdicaux et des produits vÃĐtÃĐrinaires seraient touchÃĐs par le rÃĻglement proposÃĐ.
à lâaide de donnÃĐes internes, SantÃĐ Canada a estimÃĐ quâenviron 26 % de tous les demandeurs ou fabricants touchÃĐs rÃĐpondraient à la dÃĐfinition de petites entreprises au Canada. La plus grande proportion de petites entreprises dans leur catÃĐgorie de produits est celle des instruments mÃĐdicaux (36 %) et des mÃĐdicaments vÃĐtÃĐrinaires (27 %).
Bien quâil nây aurait pas dâexemptions ou de processus particuliers proposÃĐs dans le nouveau rÃĻglement pour les petites entreprises, en dehors de ce qui existe dÃĐjà dans la rÃĐglementation et dans la pratique, les besoins des petites entreprises ont ÃĐtÃĐ pris en considÃĐration lors de la crÃĐation du rÃĻglement. Les petites entreprises bÃĐnÃĐficieraient ÃĐgalement des flexibilitÃĐs et de la clartÃĐ accrues fournies par la modification proposÃĐe. Par exemple, les petites entreprises pharmaceutiques pourraient bÃĐnÃĐficier de la flexibilitÃĐ offerte par lâautorisation prÃĐcoce des drogues dans le cadre de lâoption dâun examen en continu, ou attribuer leurs ressources plus efficacement, car les modifications proposÃĐes apporteraient plus de clartÃĐ. En gÃĐnÃĐral, de nombreuses dispositions prendraient en considÃĐration les coÃŧts de conformitÃĐ et dâadministration ÃĐventuels et/ou les consÃĐquences dâÃĐvÃĐnements indÃĐsirables avant dâimposer des conditions ou un PGR à un demandeur ou à un fabricant.
- Nombre de petites entreprises touchÃĐes : 225
- Nombre dâannÃĐes : 10 (de 2024 Ã 2033)
- AnnÃĐe de rÃĐfÃĐrence pour lâÃĐtablissement des coÃŧts : 2021
- AnnÃĐe de rÃĐfÃĐrence de la VA : 2024
- Taux dâactualisation : 7 %
| ActivitÃĐ | Valeur annualisÃĐe | Valeur actualisÃĐe |
|---|---|---|
| Conditions | 4 471 413 $ | 31 405 337 $ |
| PGR | 169 330 $ | 1 189 304 $ |
| Examens en continu | 11 030 $ | 77 470 $ |
| CoÃŧt total de la conformitÃĐ | 4 651 774 $ | 32 672 112 $ |
| CoÃŧt par petite entreprise touchÃĐe | 20 675 $ | 145 209 $ |
RÃĻgle du ÂŦ un pour un Âŧ
La rÃĻgle du ÂŦ un pour un Âŧ sâapplique puisquâil y aurait une diminution progressive du fardeau administratif pour les entreprises et le projet serait considÃĐrÃĐ comme une rÃĐduction du fardeau administratif en vertu de la rÃĻgle. La modification proposÃĐe aux exigences relatives aux normes permettrait aux fabricants de drogues dâÃĐconomiser le temps consacrÃĐ Ã la surveillance des pharmacopÃĐes de lâannexe B et à lâÃĐvaluation des rÃĐpercussions. Les ressources requises par les entreprises pour surveiller, ÃĐvaluer les impacts et informer SantÃĐ Canada lorsque les normes ont changÃĐ seraient considÃĐrÃĐes comme un fardeau administratif. Ces activitÃĐs exigent des fabricants quâils consacrent en moyenne 37,5 heures à chaque publication de pharmacopÃĐe au tarif de 31 $ de lâheurerÃĐfÃĐrence 21, et on suppose quâil y a trois publications par annÃĐe. La rÃĐduction annuelle du fardeau administratif prÃĐvu pour les entreprises est estimÃĐe à 25 177 $ (dollars de 2012) ou à 1 678 $ (dollars de 2012) par entreprise.
Lâimposition de conditions aurait pour objectif de gÃĐrer les incertitudes relatives aux avantages et/ou risques dâun produit au moment de lâautorisation ou de gÃĐrer les risques ou incertitudes ÃĐmergents aprÃĻs lâautorisation. Cette exigence serait directement liÃĐe à la protection de la santÃĐ et de la sÃĐcuritÃĐ des Canadiens. Par consÃĐquent, les coÃŧts liÃĐs au respect des conditions ne sont pas considÃĐrÃĐs comme un fardeau administratif tel quâil est dÃĐfini par la Loi sur la rÃĐduction de la paperasse, car leur objectif principal nâest pas dâassurer la conformitÃĐ.
Actuellement, les fabricants soumettent volontairement des PGR sur demande ainsi que des rÃĐsumÃĐs de PGR sâils sont disponibles. Ainsi, on ne sâattend pas à ce que le projet de rÃĻglement engendre des exigences administratives supplÃĐmentaires en ce qui concerne les PGR.
CoopÃĐration et harmonisation en matiÃĻre de rÃĐglementation
Bien que ces modifications rÃĐglementaires proposÃĐes ne fassent pas partie dâun plan officiel de coopÃĐration rÃĐglementaire avec un organisme de rÃĐglementation ÃĐtranger, elles harmoniseraient davantage les exigences de SantÃĐ Canada avec celles dâautres administrations comme les Ãtats-Unis, lâUnion europÃĐenne et lâAustralie.
Ãtats-Unis
La FDA des Ãtats-Unis a la capacitÃĐ dâobliger, par la loi, un titulaire dâautorisation à mener des activitÃĐs sur mesure pour gÃĐrer les risques et assurer les avantages de ses drogues une fois sur le marchÃĐ, comme les modifications proposÃĐes qui permettraient au ministre dâimposer des conditions sur toute drogue.
La FDA des Ãtats-Unis a la capacitÃĐ dâimposer des exigences post-approbation à certaines classes dâinstruments mÃĐdicaux. Ces conditions peuvent Être liÃĐes à des restrictions de vente, à des rapports pÃĐriodiques, à des exigences dâÃĐtiquetage ou à des essais, semblables aux modifications proposÃĐes concernant les conditions.
Dans le cas de certaines drogues, la FDA des Ãtats-Unis peut obliger, par la loi, la prÃĐsentation dâun plan dÃĐcrivant les stratÃĐgies dâÃĐvaluation et dâattÃĐnuation des risques [ÂŦ Risk Evaluation and Mitigation Strategies (REMS) Âŧ (disponible en anglais seulement)] du fabricant. Les REMS sont semblables aux PGR; par consÃĐquent, ces modifications proposÃĐes au RAD permettraient de mieux harmoniser les exigences canadiennes en matiÃĻre de stratÃĐgies dâattÃĐnuation des risques avec celles de la FDA des Ãtats-Unis.
La FDA des Ãtats-Unis permet lâutilisation dâexamens en continu pour certaines drogues à condition quâelles rÃĐpondent aux critÃĻres dÃĐcrits dans ses programmes de procÃĐdure accÃĐlÃĐrÃĐe, de thÃĐrapie innovatrice, dâapprobation accÃĐlÃĐrÃĐe, or dâexamen prioritaire [ÂŦ Fast Track, Breakthrough Therapy, Accelerated Approval, Priority Review Âŧ (disponible en anglais seulement)]. Les critÃĻres dâinclusion comprennent une drogue qui pourrait Être utilisÃĐe pour rÃĐpondre à une urgence de santÃĐ publique, ou une drogue ayant des preuves cliniques prÃĐliminaires indiquant quâil peut sâagir dâune amÃĐlioration importante par rapport aux thÃĐrapies existantes pour des maladies graves. Les fabricants qui dÃĐposent des demandes dâexamen en continu auprÃĻs de la FDA des Ãtats-Unis ont des interactions plus frÃĐquentes avant la mise en marchÃĐ avec lâorganisme de rÃĐglementation et bÃĐnÃĐficient dâun accÃĻs plus rapide au marchÃĐ, ce qui serait le cas pour les fabricants qui sont admissibles à des examens en continu avec SantÃĐ Canada en vertu des modifications proposÃĐes au RAD.
Avant lâautorisation dâune prÃĐsentation de drogue nouvelle pour certaines drogues composÃĐes de petites molÃĐcules, pharmaceutiques et biologiques, la FDA des Ãtats-Unis ÃĐvalue les ÃĐtablissements au moyen dâinspections sur place prÃĐalables à lâautorisation et/ou dâexamens des dossiers dâÃĐtablissement. Ces inspections sont menÃĐes par des inspecteurs, des enquÊteurs et dâautres spÃĐcialistes des produits dâune maniÃĻre axÃĐe sur les risques, en tenant compte des risques liÃĐs aux installations, aux produits et aux processus. Lâinspection est effectuÃĐe pour contribuer à lâassurance de la FDA des Ãtats-Unis quâun ÃĐtablissement de fabrication nommÃĐ dans une prÃĐsentation de drogue est prÊt et capable de fabriquer une drogue, et que les donnÃĐes soumises sont complÃĻtes, exactes et conformes à celles du site de fabrication. Cela est analogue à la pratique de SantÃĐ Canada de mener des ESP et à la modification proposÃĐe qui permettrait de recueillir les documents ou les renseignements à lâappui de lâÃĐvaluation dâune prÃĐsentation de drogue nouvelle.
La FDA des Ãtats-Unis exige des fabricants quâils prÃĐsentent des donnÃĐes sur lâinnocuitÃĐ et lâefficacitÃĐ par certaines sous-populations (par exemple par ÃĒge, par sexe, par race ou origine ethnique) et, dans le cas de lâefficacitÃĐ des donnÃĐes, les fabricants doivent indiquer toute modification à la posologie nÃĐcessaire pour ces sous-populations prÃĐcises.
La FDA des Ãtats-Unis exige quâune drogue soit conforme à la norme de la ÂŦ United States Phamacopeia/National Formulary (USP/NF) Âŧ si la drogue figure dans cette pharmacopÃĐe, à moins que certaines conditions ne soient remplies. Elle nâexige pas quâune liste de pharmacopÃĐes soit surveillÃĐe. GrÃĒce à ces modifications proposÃĐes, les fabricants qui revendiquent une norme dâun fabricant pour leur drogue autorisÃĐe au Canada nâauraient plus besoin de surveiller une liste de pharmacopÃĐes et de mettre à jour leur produit, au besoin. La FDA des Ãtats-Unis nâexige pas quâun fabricant inclue la norme dâune drogue sur son ÃĐtiquette. Les modifications proposÃĐes seraient conformes à cette pratique.
Union europÃĐenne
LâEMA de lâUnion europÃĐenne a la capacitÃĐ dâobliger, par la loi, un titulaire dâautorisation à mener des activitÃĐs sur mesure pour gÃĐrer les risques et assurer les avantages de ses drogues une fois sur le marchÃĐ, comme les modifications proposÃĐes qui permettraient au ministre dâimposer des conditions sur toutes les drogues. LâEMA peut ÃĐgalement imposer des conditions sur les instruments mÃĐdicaux dans des cas exceptionnels liÃĐs à la santÃĐ publique ou à la sÃĐcuritÃĐ des patients.
Comme il est indiquÃĐ dans leur ligne directrice sur les PGR (disponible en anglais seulement), lâEMA exige des fabricants quâils soumettent des PGR lorsquâils demandent une autorisation de mise sur le marchÃĐ qui sont proportionnels aux risques cernÃĐs et aux risques potentiels du produit mÃĐdicinal, ainsi quâà la nÃĐcessitÃĐ de disposer de donnÃĐes de sÃĐcuritÃĐ aprÃĻs lâautorisation. LâEMA publie ÃĐgalement des rÃĐsumÃĐs de PGR pour les drogues autorisÃĐes centralement. Les modifications proposÃĐes au RAD permettraient, par consÃĐquent, de mieux harmoniser les exigences canadiennes en matiÃĻre de stratÃĐgies dâattÃĐnuation des risques avec celles de lâEMA en permettant que le PGR et les rÃĐsumÃĐs soient prÃĐsentÃĐs dans le format de lâEMA.
LâEMA permet de dÃĐposer des prÃĐsentations aux fins dâexamen en continu dans des situations dâurgence comme la pandÃĐmie de COVID-19.
LâEMA exige que les fabricants prÃĐsentent des renseignements sur les populations de sujets dâÃĐtude dÃĐsagrÃĐgÃĐs selon lâÃĒge et le sexe et quâils fournissent une justification lorsque ces donnÃĐes ne sont pas disponibles.
LâEMA exige quâune drogue soit conforme aux normes europÃĐennes en matiÃĻre de pharmacopÃĐe lorsquâil en existe une. Elle nâexige pas quâune liste de pharmacopÃĐes soit surveillÃĐe. GrÃĒce aux modifications proposÃĐes, les fabricants qui revendiquent une norme dâun fabricant pour leur drogue autorisÃĐe au Canada nâauraient plus besoin de surveiller une liste de pharmacopÃĐes et de mettre à jour leur produit, au besoin.
LâEMA nâexige pas quâun fabricant inclue la norme dâune drogue sur son ÃĐtiquette. Les modifications proposÃĐes seraient conformes à cette pratique.
Royaume-Uni
La Medicines and Healthcare products Regulatory Agency (MHRA) du Royaume-Uni a la capacitÃĐ dâobliger, par la loi, un titulaire dâautorisation à mener des activitÃĐs sur mesure pour gÃĐrer les risques et assurer les avantages de ses drogues une fois sur le marchÃĐ, comme les modifications proposÃĐes qui permettraient au ministre dâimposer des conditions sur toutes les drogues.
La MHRA exige que les promoteurs soumettent des PGR, des rÃĐsumÃĐs et des mises à jour de PGR pour les nouvelles drogues. Elle continue dâaccepter les versions de lâUnion europÃĐenne dâun PGR, mais peut ÃĐgalement exiger une annexe comprenant du contenu national.
La MHRA a publiÃĐ ses lignes directrices sur les prÃĐsentations dâexamens en continu (disponible en anglais seulement) en dÃĐcembre 2020 dans le cadre de sa politique Brexit visant à simplifier lâÃĐlaboration de nouvelles drogues, y compris les nouvelles substances actives ainsi que les biosimilaires.
Australie
La Therapeutic Goods Administration (TGA) de lâAustralie a la capacitÃĐ dâimposer des conditions au moment de la dÃĐlivrance dâun certificat dâÃĐvaluation de la conformitÃĐ pour un instrument mÃĐdical, ou aprÃĻs la dÃĐlivrance du certificat, semblable aux modifications proposÃĐes qui permettraient au ministre dâimposer des conditions sur lâhomologation dâinstruments mÃĐdicaux.
La TGA exige des PGR, des rÃĐsumÃĐs et des mises à jour pour des produits spÃĐcifiÃĐs (à risque plus ÃĐlevÃĐ). Elle continue dâaccepter les versions de lâUnion europÃĐenne dâun PGR, mais peut ÃĐgalement exiger une annexe comprenant du contenu national.
La TGA exige que, lorsquâune norme est disponible pour une drogue dans lâune des pharmacopÃĐes par dÃĐfaut dÃĐcrites dans leur lÃĐgislation, une drogue doit au minimum satisfaire à ces normes. Les exceptions sont examinÃĐes au cas par cas. Avec la modification proposÃĐe au RAD, les normes du fabricant qui diffÃĻrent de celles des pharmacopÃĐes ÃĐnumÃĐrÃĐes à lâannexe B seraient permises, si elles ÃĐtaient acceptables par le ministre. Les limites devraient Être nuancÃĐes par des renseignements scientifiques aux fins dâexamen par le ministre dans le cadre dâune prÃĐsentation de drogue.
La TGA nâexige pas quâun fabricant inclue la norme dâune drogue sur son ÃĐtiquette. Les modifications proposÃĐes seraient conformes à cette pratique.
Harmonisation avec dâautres programmes internationaux
Les modifications apportÃĐes au RIM harmonisent les exigences canadiennes avec les lignes directrices dÃĐcrites par le Forum international des organismes de rÃĐglementation des matÃĐriels mÃĐdicaux (IMDRF) en continuant de mettre lâaccent sur lâinnocuitÃĐ et lâefficacitÃĐ de lâinstrument.
SantÃĐ Canada est membre de lâInternational Council for Harmonisation. Le Conseil a ÃĐlaborÃĐ une ligne directrice (E2E) dÃĐcrivant les ÃĐlÃĐments fondamentaux des PGR que le Canada a adoptÃĐe en 2009. Par la suite, le Canada a publiÃĐ la ligne directrice nationale connexe en 2015. Ã part les opioÃŊdes, le systÃĻme canadien ne repose actuellement que sur la ligne directrice, tandis que dâautres administrations importantes (par exemple lâEurope, lâAustralie) ont des rÃĻglements en place.
Dâautres pays comme lâAustralie, Singapour, la Suisse et le Royaume-Uni permettent le dÃĐpÃīt de prÃĐsentations en vue dâun examen en continu dans des situations dâurgence.
SantÃĐ Canada a utilisÃĐ des examens en continu pour permettre des examens conjoints de mÃĐdicaments vÃĐtÃĐrinaires avec la FDA des Ãtats-Unis dans le cadre du Conseil de coopÃĐration en matiÃĻre de rÃĐglementation. Les modifications rÃĐglementaires proposÃĐes codifieraient cette pratique.
En ce qui a trait à lâapproche pour les produits biologiques, le Canada est semblable à dâautres administrations (les Ãtats-Unis, lâUnion europÃĐenne) et à lâOrganisation mondiale de la santÃĐ en ce sens quâil a des rÃĻgles propres aux produits. Les rÃĻgles propres aux produits du Canada sont trop prescriptives et dÃĐsuÃĻtes. Les modifications rÃĐglementaires proposÃĐes offriraient la souplesse nÃĐcessaire pour suivre le rythme de lâÃĐvolution des sciences et de la technologie au fil du temps grÃĒce à des rÃĻglements axÃĐs sur les rÃĐsultats. SantÃĐ Canada continuerait de se fier aux lignes directrices internationales, comme celles ÃĐtablies par le Conseil.
Ãvaluation environnementale stratÃĐgique
ConformÃĐment à la Directive du Cabinet sur lâÃĐvaluation environnementale des projets de politiques, de plans et de programmes, une analyse prÃĐliminaire a ÃĐtÃĐ effectuÃĐe qui a permis de conclure quâil nây a pas dâeffets environnementaux importants prÃĐvus, positifs ou nÃĐgatifs; par consÃĐquent, une analyse dÃĐtaillÃĐe nâest pas justifiÃĐe.
Analyse comparative entre les sexes plus
SantÃĐ Canada sâattend à ce que ce projet ait une incidence positive sur toutes les personnes au Canada. Dans certains cas, la sous-reprÃĐsentation dans les essais cliniques et la non-disponibilitÃĐ des donnÃĐes dans lâespace post-commercialisation pourraient faire en sorte que des sous-populations prÃĐcises soient confrontÃĐes à des risques disproportionnÃĐs lorsquâelles utilisent des instruments mÃĐdicaux et des drogues en particulier. Selon les drogues et les instruments mÃĐdicaux qui sont autorisÃĐs et la façon dont les modifications rÃĐglementaires proposÃĐes sont mises en Åuvre, il est possible que certaines personnes au Canada en retirent un plus grand avantage et quâil soit possible de surmonter certains obstacles auxquels sont confrontÃĐes les populations à la recherche dâÃĐquitÃĐ et les populations dÃĐtentrices de droits (par exemple les femmes et les personnes de diverses identitÃĐs de genre, les minoritÃĐs raciales et ethniques, les personnes handicapÃĐes, les PremiÃĻres Nations, les Inuits et les MÃĐtis, etc.).
Conditions pour les instruments mÃĐdicaux
Le nombre de personnes handicapÃĐes augmente considÃĐrablement en raison dâune population vieillissante et de lâaugmentation des conditions de santÃĐ chroniquerÃĐfÃĐrence 22,rÃĐfÃĐrence 23. Ãtant donnÃĐ que lâutilisation dâinstruments mÃĐdicaux chez les personnes handicapÃĐes est disproportionnellement ÃĐlevÃĐe comparativement à la population canadienne en gÃĐnÃĐralrÃĐfÃĐrence 24, on sâattend à ce que les personnes handicapÃĐes bÃĐnÃĐficient de façon disproportionnÃĐe de la modification proposÃĐe aux conditions des instruments mÃĐdicaux. Reconnaissant lâintersectionnalitÃĐ, lâinvaliditÃĐ peut Être plus rÃĐpandue dans certaines sous-populations, avec une prÃĐvalence plus ÃĐlevÃĐe apparaissant chez les personnes ÃĒgÃĐes, chez les femmesrÃĐfÃĐrence 25 et en particulier chez les femmes autochtonesrÃĐfÃĐrence 26,rÃĐfÃĐrence 27.
De plus, la race, le sexe et lâÃĒge peuvent, dans certains cas, influencer lâinnocuitÃĐ ou lâefficacitÃĐ dâun instrument mÃĐdical. Parfois, certains sous-groupes de la population sont sous-reprÃĐsentÃĐs dans la conception dâun instrument mÃĐdical et dans les ÃĐtudes cliniques ou les essais expÃĐrimentaux qui y sont associÃĐs, ce qui pourrait prÃĐsenter des difficultÃĐs à cerner toute diffÃĐrence dans lâinnocuitÃĐ ou lâefficacitÃĐ dâun instrument mÃĐdical dans ces populations. Par exemple, certaines publications rÃĐcentes ont indiquÃĐ que les sphygmo-oxymÃĻtres peuvent donner des lectures biaisÃĐes des niveaux dâoxygÃĻne du sang sur la peau plus sombre. Les patients noirs ÃĐtaient 29 % moins susceptibles que les patients blancs de voir leur besoin de traitement reconnu par le lecteur dâoxygÃĻnerÃĐfÃĐrence 28.
Les modifications proposÃĐes aux conditions rÃĐgissant les instruments mÃĐdicaux devraient faire augmenter les niveaux de confiance envers les instruments mÃĐdicaux utilisÃĐs par les diverses populations au Canada.
DonnÃĐes dÃĐsagrÃĐgÃĐes, conditions et PGR pour les drogues
Fournir aux organismes de rÃĐglementation des donnÃĐes cliniques dÃĐsagrÃĐgÃĐes par sous-groupes de population est une ÃĐtape nÃĐcessaire pour dÃĐterminer les diffÃĐrences potentielles dans lâefficacitÃĐ et lâinnocuitÃĐ dâune drogue au sein de diverses populations. Il est de plus en plus ÃĐvident que lâinnocuitÃĐ et lâefficacitÃĐ de certains produits de santÃĐ sont touchÃĐes par un certain nombre de variables, y compris, entre autres facteurs, le sexe, lâÃĒge et la race. Par exemple, certaines drogues sont mÃĐtabolisÃĐes à diffÃĐrents degrÃĐs par les populations masculine et fÃĐminine (par exemple AmbienrÃĐfÃĐrence 29) et par diffÃĐrents groupes raciaux partageant des traits gÃĐnÃĐtiques spÃĐcifiques (par exemple le clopidogrelrÃĐfÃĐrence 30, la warfarinerÃĐfÃĐrence 31). Cela peut entraÃŪner des risques accrus pour ces sous-groupes de population prÃĐcis, contribuant ainsi aux inÃĐgalitÃĐs en matiÃĻre de santÃĐ dans le secteur des soins de santÃĐ.
De plus, certaines populations de sous-groupes demeurent sous-reprÃĐsentÃĐes dans les essais cliniques, comme les femmes enceintes, les personnes vivant dans les communautÃĐs rurales, et les personnes ÃĒgÃĐes, ce qui rend difficile lâidentification des principales diffÃĐrences en matiÃĻre dâinnocuitÃĐ ou dâefficacitÃĐ des drogues au sein de celles-ci.
La rÃĐception de donnÃĐes ventilÃĐes par sous-groupes de population (par exemple ÃĒge, sexe, race) permettrait à SantÃĐ Canada dâÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de la nouvelle drogue contre ces populations sous-reprÃĐsentÃĐes, le cas ÃĐchÃĐant. Les modifications proposÃĐes permettraient ÃĐgalement au MinistÃĻre dâexiger lâanalyse, dans les spÃĐcifications dâinnocuitÃĐ, de la rÃĐpartition des risques et des incertitudes entre les sous-populations concernÃĐes. Le cas ÃĐchÃĐant, le ministre pourrait demander des mesures à inclure dans un PGR. Ces mesures et les renseignements sur lâinnocuitÃĐ reçus par le MinistÃĻre à la suite de ces mesures pourraient Être pris en considÃĐration par le ministre dans sa surveillance continue aprÃĻs la mise en marchÃĐ de lâinnocuitÃĐ de la drogue. Les mesures peuvent comprendre la façon dont les donnÃĐes seraient recueillies auprÃĻs de diverses populations de patients. De plus, les conditions pourraient Être utilisÃĐes pour exiger que des donnÃĐes supplÃĐmentaires soient recueillies aprÃĻs lâautorisation en ce qui concerne les sous-populations de patients pour lesquelles il y a des donnÃĐes cliniques limitÃĐes et oÃđ lâinnocuitÃĐ ou lâefficacitÃĐ de la drogue en question doit Être mieux caractÃĐrisÃĐe. Les modifications proposÃĐes constitueraient des leviers importants pour promouvoir une prise en considÃĐration accrue de diverses sous-populations dans les essais cliniques et pour la collecte de donnÃĐes dÃĐsagrÃĐgÃĐes supplÃĐmentaires durant la post-commercialisation.
Examens en continu
Les deux tiers des personnes touchÃĐes par des maladies rares sont des enfants. Parce quâelles touchent un nombre relativement petit de patients, il y a une comprÃĐhension limitÃĐe en ce qui concerne leur diagnostic et leur traitementrÃĐfÃĐrence 32. Lâoption proposÃĐe pour les examens en continu pourrait contribuer à lâapprobation prÃĐcoce des drogues pour le traitement de telles maladies.
De plus, bien que tout le monde soit susceptible aux maladies infectieuses, certaines populations sont vulnÃĐrables à des rÃĐsultats plus graves (par exemple les adultes plus ÃĒgÃĐs, les personnes atteintes de maladies chroniques et les personnes de tout ÃĒge qui sont immunodÃĐprimÃĐes)rÃĐfÃĐrence 33. Lâoption dâexamen en continu proposÃĐ pourrait contribuer à lâautorisation prÃĐcoce des drogues qui traitent une maladie infectieuse ÃĐmergente et, par consÃĐquent, profiterait à toutes les personnes au Canada, mais en particulier aux sous-populations qui sont les plus susceptibles de rÃĐsultats graves et à celles qui sont plus susceptibles dâÊtre exposÃĐes à des maladies infectieuses.
Normes et modernisation des exigences relatives aux produits biologiques
On ne sâattend pas à ce que les changements apportÃĐs aux normes aient une incidence sur les sous-populations. De plus, ÃĐtant donnÃĐ que les modifications proposÃĐes pour moderniser le rÃĻglement sur les produits biologiques appuient les pratiques actuelles, on ne sâattend pas à ce quâelles aient une incidence sur la population en gÃĐnÃĐral.
Mise en Åuvre, conformitÃĐ et application, et normes de service
Mise en Åuvre
Les modifications rÃĐglementaires proposÃĐes suivantes, qui accordent la prioritÃĐ Ã la rÃĐduction du fardeau pour lâindustrie et garantissent que SantÃĐ Canada est bien placÃĐ pour faire face à toute future urgence de santÃĐ publique, entreraient en vigueur au moment de lâenregistrement :
- les modifications relatives aux drogues dâurgence de santÃĐ publique (y compris les conditions, les examens en continu et le prÃĐpositionnement);
- assurer la qualitÃĐ des drogues pendant la fabrication;
- la modernisation des exigences concernant les produits biologiques;
- les renseignements à lâappui de lâexamen des prÃĐsentations de drogue;
- les donnÃĐes dÃĐsagrÃĐgÃĐes relatives aux essais cliniques pour les prÃĐsentations de drogues nouvelles pour usage humain et les supplÃĐments à une prÃĐsentation de drogue nouvelle pour usage humain;
- les normes.
Les modifications rÃĐglementaires proposÃĐes suivantes, qui nÃĐcessitent un dÃĐlai supplÃĐmentaire pour Être mises en Åuvre, entreraient en vigueur un an aprÃĻs lâenregistrement :
- les conditions (pour toutes les drogues et les instruments mÃĐdicaux de classe II, III et IV);
- les plans de gestion des risques;
- les examens en continu (pour toutes les drogues sauf les drogues dâurgence de santÃĐ publique).
La modification rÃĐglementaire proposÃĐe suivante entrerait en vigueur à un moment fixÃĐ dans une future modification rÃĐglementaire une fois que le MinistÃĻre aura dÃĐterminÃĐ que les dispositions propres à la COVID-19 ne sont plus nÃĐcessaires :
- modifier la dÃĐfinition de drogue dâurgence de santÃĐ publique pour ne plus inclure une nouvelle drogue dont lâusage ou les conditions dâemploi recommandÃĐs par le fabricant se rapportent à la COVID-19;
- abroger dâautres dispositions spÃĐcifiques à la COVID-19.
Le MinistÃĻre prÃĐvoit mener des consultations sur les principales lignes directrices mises à jour qui accompagneraient le rÃĻglement proposÃĐ Ã peu prÃĻs en mÊme temps que la publication prÃĐalable du rÃĻglement dans la Partie I de la Gazette du Canada. SantÃĐ Canada publierait les documents dâorientation mis à jour au fur et à mesure quâils seraient finalisÃĐs sur Canada.ca.
Les lignes directrices fourniraient aux fabricants les dÃĐtails sur la façon dont ils peuvent se conformer aux nouvelles dispositions proposÃĐes.
ConformitÃĐ et application
La conformitÃĐ et lâapplication du rÃĻglement proposÃĐ resteront conformes à une approche fondÃĐe sur le risque, harmonisÃĐe aux politiques ministÃĐrielles actuelles, notamment la promotion et la surveillance de la conformitÃĐ et les activitÃĐs dâapplication conformÃĐment à la Politique de conformitÃĐ et dâapplication de la loi pour les produits de santÃĐ (POL-0001) du MinistÃĻre.
SantÃĐ Canada utilise un large ÃĐventail de mesures et dâoutils de conformitÃĐ et dâapplication de la loi. Les actions, les outils et le niveau dâintervention utilisÃĐs dÃĐpendent du contexte, de la situation et du risque pour la santÃĐ. Certaines actions et certains outils sont conçus pour aider les parties rÃĐglementÃĐes à comprendre leurs responsabilitÃĐs en vertu de la loi, tandis que dâautres actions et outils sont conçus pour les inciter à se conformer à la loi. Au besoin, on utilisera des mesures dâexÃĐcution pour remÃĐdier au non-respect de la loi. Par exemple, le dÃĐfaut de se conformer aux conditions constituerait une infraction à lâarticle 21.7 de la Loi et pourrait entraÃŪner le MinistÃĻre à prendre des mesures de conformitÃĐ et dâapplication de la loi conformÃĐment à la POL-0001 ou à renvoyer lâaffaire au Service des poursuites pÃĐnales du Canada en vue dâune poursuite ÃĐventuelle.
Personne-ressource
Bruno Rodrigue
Directeur exÃĐcutif
Bureau de la modernisation des lois et des rÃĻglements
Direction des politiques, de la planification et des affaires internationales
Direction gÃĐnÃĐrale des produits de santÃĐ et des aliments
SantÃĐ Canada
Holland Cross, bureau P2108
11, avenue Holland
Ottawa (Ontario)
K1A 0K9
Indice de lâadresse : 3000A
Courriel : lrm.consultations-mlr@hc-sc.gc.ca
PROJET DE RÃGLEMENTATION
Avis est donnÃĐ que la gouverneure en conseil, en vertu de lâarticle 30rÃĐfÃĐrence a de la Loi sur les aliments et drogues rÃĐfÃĐrence b, se propose de prendre le RÃĻglement modifiant certains rÃĻglements pris en vertu de la Loi sur les aliments et drogues (homologation agile), ci-aprÃĻs.
Les intÃĐressÃĐs peuvent prÃĐsenter leurs observations au sujet du projet de rÃĻglement dans les cent jours suivant la date de publication du prÃĐsent avis. Ils sont fortement encouragÃĐs à le faire au moyen de lâoutil en ligne disponible à cet effet sur le site Web de la Gazette du Canada. Sâils choisissent plutÃīt de prÃĐsenter leurs observations par courriel, par la poste ou par tout autre moyen, ils sont priÃĐs dây citer la Partie I de la Gazette du Canada, ainsi que la date de publication du prÃĐsent avis, et dâenvoyer le tout à Bruno Rodrigue, directeur exÃĐcutif, Bureau de la modernisation des lois et des rÃĻglements, Direction gÃĐnÃĐrale des produits de santÃĐ et des aliments, ministÃĻre de la SantÃĐ, indice dâadresse 3000A, 11, avenue Holland, bureau P2108, Ottawa (Ontario) K1A 0K9 (courriel : lrm.consultations-mlr@hc-sc.gc.ca).
Ottawa, le 6 dÃĐcembre 2022
La greffiÃĻre adjointe du Conseil privÃĐ
Wendy Nixon
RÃĻglement modifiant certains rÃĻglements pris en vertu de la Loi sur les aliments et drogues (homologation agile)
RÃĻglement sur les aliments et drogues
1 LâalinÃĐa A.01.048d) du RÃĻglement sur les aliments et drogues rÃĐfÃĐrence 34 est remplacÃĐ par ce qui suit :
- d) les articles C.04.001 Ã C.04.006.
2 (1) Les sous-alinÃĐas C.01.004(1)a)(iii) et (iv) du mÊme rÃĻglement sont abrogÃĐs.
(2) Le paragraphe C.01.004(5) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(5) Les paragraphes (1) Ã (3) ne sâappliquent pas :
- a) aux drogues vendues à un fabricant de drogues;
- b) aux drogues vendues sur ordonnance dont lâÃĐtiquette porte le mode dâemploi appropriÃĐ de la drogue et est conforme aux exigences de lâarticle C.01.005;
- c) aux drogues visÃĐes à lâannexe D de la Loi.
3 Lâarticle C.01.011 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (4), de ce qui suit :
(5) Le paragraphe (4) ne sâapplique pas aux drogues nouvelles autres que celles qui figurent à lâannexe C de la Loi.
4 Lâarticle C.01.014.1 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (3), de ce qui suit :
(4) Le ministre peut demander au fabricant dâune drogue pour usage humain — autre quâune drogue nouvelle — qui a prÃĐsentÃĐ une demande en vertu du paragraphe (1) à lâÃĐgard de la drogue de lui fournir un plan de gestion des risques conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de cette derniÃĻre sâil a des motifs raisonnables de croire :
- a) soit que le degrÃĐ dâincertitude quant aux risques associÃĐs à la drogue est considÃĐrable;
- b) soit que la drogue prÃĐsente un risque grave de prÃĐjudice à la santÃĐ humaine qui justifie la prise de mesures — autres que lâÃĐtiquetage — visant à diminuer la probabilitÃĐ quâun tel prÃĐjudice survienne ou à en amoindrir la gravitÃĐ.
5 Le paragraphe C.01.014.2(2) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(2) Le ministre peut refuser de dÃĐlivrer le document dans les cas suivants :
- a) il a des motifs raisonnables de croire que le produit faisant lâobjet de la demande dâidentification numÃĐrique nâest pas une drogue;
- b) il a des motifs raisonnables de croire que la vente de la drogue :
- (i) soit causerait un prÃĐjudice à la santÃĐ de lâacheteur ou du consommateur,
- (ii) soit contreviendrait à la Loi ou au prÃĐsent rÃĻglement;
- c) dans le cas oÃđ il a demandÃĐ au fabricant de la drogue, en vertu du paragraphe C.01.014.1(4), de lui fournir un plan de gestion des risques à lâÃĐgard de celle-ci, ce dernier ne lui a pas fourni un plan de gestion des risques conforme aux exigences de lâarticle C.01.700.
6 (1) Le paragraphe C.01.014.21(1.1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(1.1) Le ministre peut, en tout temps, assortir de conditions lâidentification numÃĐrique attribuÃĐe à une drogue dâurgence de santÃĐ publique ou modifier ces conditions dans les cas suivants :
- a) un avis de conformitÃĐ a ÃĐtÃĐ dÃĐlivrÃĐ en vertu de lâarticle C.08.004 à lâÃĐgard :
- (i) soit dâune prÃĐsentation de drogue nouvelle qui a ÃĐtÃĐ dÃĐposÃĐe au titre de lâarticle C.08.002 à lâÃĐgard de la drogue dâurgence de santÃĐ publique et qui contient la mention prÃĐvue à lâalinÃĐa C.08.002(2.1)a),
- (ii) soit dâun supplÃĐment à une telle prÃĐsentation de drogue nouvelle qui a ÃĐtÃĐ dÃĐposÃĐ au titre de lâarticle C.08.003 à lâÃĐgard de la drogue dâurgence de santÃĐ publique;
- b) un avis de conformitÃĐ a ÃĐtÃĐ dÃĐlivrÃĐ en vertu de lâarticle C.08.004 à lâÃĐgard de lâune des prÃĐsentations ou de lâun des supplÃĐments ci-aprÃĻs qui a ÃĐtÃĐ dÃĐposÃĐ Ã lâÃĐgard de la drogue sur la base dâune comparaison directe ou indirecte entre celle-ci et une autre drogue dâurgence de santÃĐ publique visÃĐe à lâalinÃĐa a) :
- (i) la prÃĐsentation de drogue nouvelle visÃĐe à lâarticle C.08.002,
- (ii) la prÃĐsentation abrÃĐgÃĐe de drogue nouvelle visÃĐe à lâarticle C.08.002.1,
- (iii) le supplÃĐment à une prÃĐsentation de drogue nouvelle ou à une prÃĐsentation abrÃĐgÃĐe de drogue nouvelle visÃĐ Ã lâarticle C.08.003.
(2) La dÃĐfinition de drogue dÃĐsignÃĐe contre la COVID-19, au paragraphe C.01.014.21(3) du mÊme rÃĻglement, est abrogÃĐe.
(3) Le paragraphe C.01.014.21(3) du mÊme rÃĻglement est modifiÃĐ par adjonction, selon lâordre alphabÃĐtique, de ce qui suit :
- drogue dâurgence de santÃĐ publique
- Sâentend au sens de lâarticle C.08.001.1. (public health emergency drug)
(4) Lâarticle C.01.014.21 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
C.01.014.21 Le ministre peut, en tout temps, assortir de conditions lâidentification numÃĐrique attribuÃĐe à une drogue ou modifier de telles conditions aprÃĻs avoir pris en compte les facteurs suivants :
- a) la question de savoir si les avantages ou les risques associÃĐs à la drogue font lâobjet dâincertitudes importantes;
- b) la question de savoir si les exigences prÃĐvues sous le rÃĐgime de la Loi sont suffisantes pour atteindre les objectifs suivants :
- (i) optimiser les avantages et gÃĐrer les risques associÃĐs à la drogue,
- (ii) gÃĐrer les incertitudes dont ces avantages et ces risques font lâobjet,
- (iii) recueillir des renseignements permettant dâÃĐvaluer en continu ces avantages et ces risques, dÃĐceler tout changement à leur ÃĐgard et gÃĐrer ces incertitudes;
- c) la question de savoir si les conditions projetÃĐes peuvent contribuer à lâatteinte des objectifs mentionnÃĐs aux sous-alinÃĐas b)(i) à (iii);
- d) la question de savoir si le respect des conditions projetÃĐes est rÃĐalisable sur le plan technique;
- e) la question de savoir sâil existe des moyens moins exigeants pour atteindre les objectifs des conditions projetÃĐes.
7 Le mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâarticle C.01.625, de ce qui suit :
Plan de gestion des risques
C.01.700 Tout plan de gestion des risques fourni au ministre sous le rÃĐgime du prÃĐsent rÃĻglement doit tenir compte du contexte canadien et comprendre ce qui suit :
- a) la description de la drogue à laquelle le plan se rapporte et de lâutilisation à laquelle cette derniÃĻre est destinÃĐe;
- b) la description dÃĐtaillÃĐe des risques associÃĐs à la drogue et des incertitudes dont ces risques font lâobjet;
- c) la description dÃĐtaillÃĐe des mesures que le fabricant entend prendre pour faire face à ces incertitudes et pour surveiller celles-ci;
- d) la description dÃĐtaillÃĐe des mesures que le fabricant entend prendre pour prÃĐvenir ou pour rÃĐduire ces risques;
- e) la description dÃĐtaillÃĐe de la façon dont le fabricant a lâintention dâÃĐvaluer lâefficacitÃĐ des mesures quâil entend prendre pour prÃĐvenir ou pour rÃĐduire ces risques;
- f) le rÃĐsumÃĐ du plan, en français et en anglais.
C.01.701 (1) Le ministre peut demander au fabricant dâune drogue pour usage humain à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe de lui fournir un plan de gestion des risques conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue si les conditions suivantes sont rÃĐunies :
- a) aucun plan de gestion des risques à lâÃĐgard de la drogue ne lui a encore ÃĐtÃĐ fourni;
- b) il a des motifs raisonnables de croire :
- (i) soit que le degrÃĐ dâincertitude quant aux risques associÃĐs à la drogue est considÃĐrable,
- (ii) soit que la drogue prÃĐsente un risque grave de prÃĐjudice à la santÃĐ humaine qui justifie la prise de mesures — autres que lâÃĐtiquetage — visant à diminuer la probabilitÃĐ quâun tel prÃĐjudice survienne ou à en amoindrir la gravitÃĐ.
(2) Dans le cas oÃđ il omet de fournir au ministre un plan de gestion des risques conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue dans le dÃĐlai prÃĐcisÃĐ dans la demande, le fabricant ne peut vendre la drogue tant quâil nâa pas fourni un tel plan à ce dernier.
C.01.702 Le fabricant dâune drogue pour usage humain à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe fournit dÃĻs que possible au ministre un plan de gestion des risques à jour et conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue dans les cas suivants :
- a) les risques associÃĐs à la drogue ou les incertitudes dont ces risques font lâobjet diffÃĻrent sensiblement de ceux qui sont dÃĐcrits dans le plan existant;
- b) les mesures que le fabricant entend prendre pour faire face aux incertitudes dont les risques associÃĐs à la drogue font lâobjet et pour surveiller celles-ci ou pour prÃĐvenir ou rÃĐduire ces risques diffÃĻrent sensiblement de celles qui sont dÃĐcrites dans le plan existant.
C.01.703 (1) Le ministre peut demander au fabricant dâune drogue pour usage humain à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe de lui fournir un plan de gestion des risques à jour conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue si, compte tenu de renseignements quâil a obtenus depuis que le plan existant lui a ÃĐtÃĐ fourni, il a des motifs raisonnables de croire :
- a) soit que les risques associÃĐs à la drogue ou les incertitudes dont ces risques font lâobjet diffÃĻrent sensiblement de ceux qui sont dÃĐcrits dans le plan existant;
- b) soit que la drogue prÃĐsente un risque grave de prÃĐjudice à la santÃĐ humaine justifiant la prise de mesures qui visent à diminuer la probabilitÃĐ quâun tel prÃĐjudice survienne ou à en amoindrir la gravitÃĐ et qui diffÃĻrent sensiblement de celles qui sont dÃĐcrites dans le plan existant.
(2) Dans le cas oÃđ il omet de fournir au ministre un plan de gestion des risques à jour conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue dans le dÃĐlai prÃĐcisÃĐ dans la demande, le fabricant ne peut vendre la drogue tant quâil nâa pas fourni un tel plan à ce dernier.
8 (1) Le paragraphe C.01A.005(2) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(2) La demande de licence dâÃĐtablissement qui vise lâune ou plusieurs des activitÃĐs figurant au tableau I de lâarticle C.01A.008 à exercer à lâÃĐgard dâune catÃĐgorie de drogues figurant au tableau II de cet article qui inclut lâune ou plusieurs des drogues ci-aprÃĻs peut contenir une mention à cet ÃĐgard :
- a) une drogue contre la COVID-19;
- b) une drogue fabriquÃĐe ou vendue en vue dâÊtre utilisÃĐe relativement à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1 ou prÃĐsentÃĐe comme pouvant lâÊtre.
(2) Le paragraphe C.01A.005(2) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(2) La demande de licence dâÃĐtablissement qui vise lâune ou plusieurs des activitÃĐs figurant au tableau I de lâarticle C.01A.008 à exercer à lâÃĐgard dâune catÃĐgorie de drogues figurant au tableau II de cet article qui inclut une drogue fabriquÃĐe ou vendue en vue dâÊtre utilisÃĐe relativement à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1, ou prÃĐsentÃĐe comme pouvant lâÊtre, peut contenir une mention à cet ÃĐgard.
9 (1) Le paragraphe C.01A.006(1.1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(1.1) La demande de modification dâune licence dâÃĐtablissement qui vise lâune ou plusieurs des activitÃĐs figurant au tableau I de lâarticle C.01A.008 à exercer à lâÃĐgard dâune catÃĐgorie de drogues figurant au tableau II de cet article qui inclut lâune ou plusieurs des drogues ci-aprÃĻs peut contenir une mention à cet ÃĐgard :
- a) une drogue contre la COVID-19;
- b) une drogue fabriquÃĐe ou vendue en vue dâÊtre utilisÃĐe relativement à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1 ou prÃĐsentÃĐe comme pouvant lâÊtre.
(2) Le paragraphe C.01A.006(1.1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(1.1) La demande de modification dâune licence dâÃĐtablissement qui vise lâune ou plusieurs des activitÃĐs figurant au tableau I de lâarticle C.01A.008 à exercer à lâÃĐgard dâune catÃĐgorie de drogues figurant au tableau II de cet article qui inclut une drogue fabriquÃĐe ou vendue en vue dâÊtre utilisÃĐe relativement à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1, ou prÃĐsentÃĐe comme pouvant lâÊtre, peut contenir une mention à cet ÃĐgard.
10 (1) Le paragraphe C.01A.008(1.1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(1.1) Lorsquâil ÃĐvalue sâil a reçu les renseignements et le matÃĐriel visÃĐs aux articles C.01A.005 à C.01A.007 à lâÃĐgard de la demande visÃĐe aux paragraphes C.01A.005(2) ou C.01A.006(1.1) qui contient la mention visÃĐe à celui de ces paragraphes qui sâapplique, le ministre prend ÃĐgalement en considÃĐration le besoin en matiÃĻre de santÃĐ publique relatif à la COVID-19 ou à lâaffection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1, selon le cas.
(2) Le paragraphe C.01A.008(1.1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(1.1) Lorsquâil ÃĐvalue sâil a reçu les renseignements et le matÃĐriel visÃĐs aux articles C.01A.005 à C.01A.007 à lâÃĐgard de la demande visÃĐe aux paragraphes C.01A.005(2) ou C.01A.006(1.1) qui contient la mention visÃĐe à celui de ces paragraphes qui sâapplique, le ministre prend ÃĐgalement en considÃĐration le besoin en matiÃĻre de santÃĐ publique relatif à lâaffection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1.
(3) Lâarticle C.01A.008 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (4), de ce qui suit :
(5) Dans le cas oÃđ la demande de dÃĐlivrance ou de modification dâune licence dâÃĐtablissement contient, conformÃĐment aux paragraphes C.01A.005(2) ou C.01A.006(1.1), une mention faisant rÃĐfÃĐrence à une affection qui est ultÃĐrieurement retirÃĐe de la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1, le ministre peut nÃĐanmoins dÃĐlivrer ou modifier la licence au titre de cette demande.
11 Lâarticle C.01A.012.1 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (2), de ce qui suit :
(3) Il est entendu que le ministre peut exercer le pouvoir que lui confÃĻre le paragraphe (1) à lâÃĐgard de la licence dâÃĐtablissement jusquâà lâannulation de celle-ci, mÊme si son titulaire ne mÃĻne aucune activitÃĐ Ã lâÃĐgard de la drogue faisant lâobjet de la mention contenue dans la demande au titre de laquelle la licence a ÃĐtÃĐ dÃĐlivrÃĐe ou modifiÃĐe.
12 Lâintertitre prÃĐcÃĐdant lâarticle C.02.013 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
ContrÃīle de la qualitÃĐ
C.02.012.1 Chaque lot ou lot de fabrication dâune drogue est manufacturÃĐ, emballÃĐ-ÃĐtiquetÃĐ, analysÃĐ et entreposÃĐ, y compris durant son transport, de façon à assurer la qualitÃĐ de la drogue.
13 Le paragraphe C.02.019(4.1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(4.1) Les paragraphes (1) et (2) ne sâappliquent pas au distributeur et à lâimportateur dâune drogue contre la COVID-19 si le lot dont elle provient fait lâobjet dâune demande au titre du paragraphe C.04.007(1).
14 Lâarticle C.03.206 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
C.03.206 Les articles C.01.005 et C.04.009 ne sâappliquent pas aux constituants et aux trousses.
15 Le titre 4 de la partie C du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
TITRE 4
Drogues de lâannexe D
DÃĐfinitions
C.04.001 Les dÃĐfinitions qui suivent sâappliquent au prÃĐsent titre.
- drogue
- Drogue visÃĐe à lâannexe D de la Loi qui est sous forme posologique ou ingrÃĐdient actif pouvant Être utilisÃĐ dans la prÃĐparation dâune drogue visÃĐe à cette annexe. (drug)
- matÃĐriel dâorigine biologique :
-
- a) Tout matÃĐriel biologique obtenu ou dÃĐrivÃĐ dâun Être humain;
- b) tout animal — notamment tout insecte — ou tout matÃĐriel biologique obtenu ou dÃĐrivÃĐ dâun animal;
- c) toute plante ou tout matÃĐriel biologique obtenu ou dÃĐrivÃĐ dâune plante;
- d) tout micro-organisme — notamment toute bactÃĐrie, tout virus, tout champignon ou tout bactÃĐriophage — ou tout matÃĐriel biologique obtenu ou dÃĐrivÃĐ dâun micro-organisme. (biological source material)
- titulaire
- Sâentend, à lâÃĐgard dâune identification numÃĐrique, du fabricant à qui le ministre a dÃĐlivrÃĐ le document qui indique cette identification en application du paragraphe C.01.014.2(1). (holder)
Interdictions de vente
C.04.002 Il est interdit au distributeur et à lâimportateur dâune drogue de vendre celle-ci à moins quâelle ait ÃĐtÃĐ manufacturÃĐe, emballÃĐe-ÃĐtiquetÃĐe et analysÃĐe conformÃĐment au prÃĐsent titre.
C.04.003 Il est interdit à la personne qui a manufacturÃĐ, emballÃĐ-ÃĐtiquetÃĐ ou analysÃĐ une drogue de vendre celle-ci à moins que la personne ait accompli lâactivitÃĐ en question conformÃĐment au prÃĐsent titre.
MatÃĐriel dâorigine biologique
C.04.004 (1) Il est interdit dâutiliser du matÃĐriel dâorigine biologique pour manufacturer une drogue à moins que les conditions suivantes soient remplies :
- a) le matÃĐriel est conforme aux exigences suivantes :
- (i) il est prÃĐparÃĐ et entreposÃĐ dâune maniÃĻre qui convient à son utilisation dans la manufacture de la drogue,
- (ii) dans le cas oÃđ il provient dâun Être humain ou dâun animal, il est prÃĐlevÃĐ sous surveillance mÃĐdicale ou vÃĐtÃĐrinaire, selon le cas;
- b) la personne qui manufacture la drogue recueille les renseignements permettant la traçabilitÃĐ du matÃĐriel;
- c) tout Être humain ou tout animal duquel le matÃĐriel est prÃĐlevÃĐ — ou tout animal qui, en soit, constitue ce matÃĐriel — nâa aucune maladie qui fait en sorte que le matÃĐriel ne convient pas à cette fin.
(2) La personne qui utilise du matÃĐriel dâorigine biologique pour manufacturer une drogue fixe la pÃĐriode durant laquelle elle conserve les renseignements visÃĐs à lâalinÃĐa (1)b) en tenant compte de ce qui suit :
- a) la nature du matÃĐriel;
- b) les risques associÃĐs au matÃĐriel;
- c) la façon dont le matÃĐriel est utilisÃĐ pour manufacturer la drogue;
- d) dans le cas oÃđ le matÃĐriel est utilisÃĐ pour manufacturer une drogue sous forme posologique, lâutilisation à laquelle la drogue est destinÃĐe.
(3) La personne qui utilise du matÃĐriel dâorigine biologique pour manufacturer une drogue conserve les renseignements visÃĐs à lâalinÃĐa (1)b) pendant au moins la plus longue des pÃĐriodes suivantes :
- a) cinq ans suivant la derniÃĻre utilisation du matÃĐriel pour manufacturer la drogue;
- b) la pÃĐriode fixÃĐe conformÃĐment au paragraphe (2).
(4) Il incombe à la personne qui utilise du matÃĐriel dâorigine biologique pour manufacturer une drogue de sâassurer que le matÃĐriel est conforme aux spÃĐcifications sây rapportant qui ont ÃĐtÃĐ fournies au ministre, le cas ÃĐchÃĐant, relativement à la drogue.
PrÃĐvention de la contamination
C.04.005 (1) Il incombe à la personne qui manufacture une drogue et à celle qui emballe une drogue dans un rÃĐcipient immÃĐdiat de faire ce qui suit :
- a) isoler toute opÃĐration faite avec des agents infectieux dont la manipulation exige des prÃĐcautions particuliÃĻres, notamment des micro-organismes sporulÃĐs pathogÃĻnes;
- b) rÃĐduire le risque de contamination de tout matÃĐriel dâorigine biologique et de toute drogue dans les locaux oÃđ elle manufacture ou emballe la drogue, selon le cas, notamment en prenant des mesures visant à ce que tout individu qui a accÃĻs à la zone de manufacture ou dâemballage soit protÃĐgÃĐ contre toute infection.
(2) Il est interdit à toute personne dâutiliser dans ses locaux tout procÃĐdÃĐ de laboratoire de nature diagnostique qui nâest pas isolÃĐ des opÃĐrations visant à manufacturer, emballer-ÃĐtiqueter ou analyser des drogues.
PrÃĐparations de rÃĐfÃĐrence
C.04.006 Les prÃĐparations de rÃĐfÃĐrence qui servent à analyser la puretÃĐ ou lâactivitÃĐ dâune drogue doivent permettre de contrÃīler la qualitÃĐ de celle-ci.
Lancement de lots
C.04.007 (1) Pour ÃĐvaluer si le lot dâune drogue sous forme posologique est propre à la vente, le ministre peut demander au manufacturier, à lâemballeur-ÃĐtiqueteur ou à lâimportateur de la drogue ou au titulaire de lâidentification numÃĐrique de lui fournir des renseignements, des ÃĐchantillons de la drogue ou des ingrÃĐdients actifs de celle-ci ou tout matÃĐriel à utiliser pour effectuer des ÃĐpreuves sur les ÃĐchantillons.
(2) Il est interdit à la personne à qui le ministre demande de fournir des renseignements, des ÃĐchantillons ou du matÃĐriel en vertu du paragraphe (1), et à toute personne à qui ce dernier notifie une telle demande, de vendre toute drogue provenant du lot visÃĐ par cette derniÃĻre, à moins que le ministre lâavise que le lot est propre à la vente.
(3) Pour lâapplication du prÃĐsent article, propre à la vente signifie que le lot a ÃĐtÃĐ manufacturÃĐ, emballÃĐ-ÃĐtiquetÃĐ et analysÃĐ conformÃĐment au prÃĐsent rÃĻglement et de façon compatible avec les renseignements qui ont ÃĐtÃĐ fournis au ministre concernant la qualitÃĐ et lâinnocuitÃĐ de la drogue en question.
Rapports pÃĐriodiques sur la qualitÃĐ
C.04.008 à la demande du ministre, le titulaire de lâidentification numÃĐrique attribuÃĐe à une drogue sous forme posologique lui fournit, chaque annÃĐe ou à tout intervalle plus long prÃĐcisÃĐ par le ministre, des renseignements sur la qualitÃĐ de la drogue et des ingrÃĐdients actifs de celle-ci, y compris des renseignements sur la rÃĐgularitÃĐ des processus de manufacture et dâemballage de la drogue et des ingrÃĐdients.
Ãtiquetage
C.04.009 (1) Les renseignements ci-aprÃĻs doivent figurer sur lâespace principal des ÃĐtiquettes intÃĐrieure et extÃĐrieure dâune drogue sous forme posologique :
- a) le nom propre de la drogue, si elle en a un, lequel est inscrit immÃĐdiatement avant ou aprÃĻs la marque nominative, si elle en a une, en caractÃĻres dâune taille au moins ÃĐgale à la moitiÃĐ de celle des caractÃĻres de la marque nominative;
- b) le nom usuel de la drogue, Ã dÃĐfaut dâun nom propre;
- c) la quantitÃĐ nette de la drogue dans lâemballage;
- d) sâagissant dâune drogue qui doit Être stÃĐrile selon le prÃĐsent rÃĻglement, les mentions ÂŦ stÃĐrile Âŧ et ÂŦ sterile Âŧ;
- e) dans le cas oÃđ le matÃĐriel dâorigine biologique utilisÃĐ pour manufacturer la drogue ou ses ingrÃĐdients actifs a ÃĐtÃĐ obtenu ou dÃĐrivÃĐ dâun Être humain, une indication à cet effet;
- f) dans le cas oÃđ le matÃĐriel dâorigine biologique utilisÃĐ pour manufacturer la drogue ou ses ingrÃĐdients actifs a ÃĐtÃĐ obtenu ou dÃĐrivÃĐ dâun animal, lâespÃĻce dâorigine.
(2) Les renseignements ci-aprÃĻs doivent figurer sur un espace quelconque des ÃĐtiquettes intÃĐrieure et extÃĐrieure dâune drogue sous forme posologique :
- a) le nom du titulaire de lâidentification numÃĐrique attribuÃĐe à la drogue;
- b) lâactivitÃĐ de la drogue, sâil y a lieu;
- c) la dose recommandÃĐe de la drogue;
- d) le numÃĐro de lot de la drogue;
- e) sous rÃĐserve du paragraphe (7), la date limite dâutilisation de la drogue;
- f) sous rÃĐserve du paragraphe (3), le mode dâemploi de la drogue;
- g) sous rÃĐserve du paragraphe (4), tout autre renseignement nÃĐcessaire en vue de prÃĐvenir un prÃĐjudice à la santÃĐ humaine.
(3) LâalinÃĐa (2)f) ne sâapplique pas si le mode dâemploi de la drogue doit figurer sur lâÃĐtiquette selon les articles C.01.004.02 ou C.01.004.03.
(4) MalgrÃĐ lâalinÃĐa (2)g), dans le cas oÃđ une autre disposition du prÃĐsent rÃĻglement prÃĐvoit quâun renseignement visÃĐ Ã cet alinÃĐa doit figurer sur un espace particulier dâune ÃĐtiquette, le renseignement doit figurer sur cet espace.
(5) Les renseignements ci-aprÃĻs doivent figurer sur un espace quelconque de lâÃĐtiquette extÃĐrieure dâune drogue sous forme posologique :
- a) lâadresse du titulaire de lâidentification numÃĐrique attribuÃĐe à la drogue;
- b) la liste quantitative des agents de conservation contenus dans la drogue, le cas ÃĐchÃĐant, identifiÃĐs par leur nom propre ou, Ã dÃĐfaut, par leur nom usuel;
- c) les conditions approuvÃĐes dâentreposage de la drogue;
- d) tout gabarit ou autre renseignement nÃĐcessaires en vue de permettre lâentreposage et la manutention convenables de la drogue;
- e) dans le cas dâune drogue nouvelle pour usage exceptionnel à lâÃĐgard de laquelle un avis de conformitÃĐ a ÃĐtÃĐ dÃĐlivrÃĐ en application de lâarticle C.08.004.01, la mention suivante, inscrite en majuscules et de façon lisible :
ÂŦ SANTà CANADA A AUTORISà LA VENTE DE CETTE DROGUE NOUVELLE POUR USAGE EXCEPTIONNEL AUX FINS DE [indication de la fin] EN SE FONDANT SUR DES ESSAIS CLINIQUES RESTREINTS CHEZ LâÃTRE HUMAIN.
HEALTH CANADA HAS AUTHORIZED THE SALE OF THIS EXTRAORDINARY USE NEW DRUG FOR [naming purpose] BASED ON LIMITED CLINICAL TESTING IN HUMANS. Âŧ.
(6) Dans le cas oÃđ le rÃĐcipient immÃĐdiat dâune drogue sous forme posologique est trop petit pour avoir une ÃĐtiquette intÃĐrieure conforme aux exigences du prÃĐsent rÃĻglement, les exigences de ce dernier relatives à lâÃĐtiquette intÃĐrieure ne sâappliquent pas si les conditions suivantes sont rÃĐunies :
- a) il y a une ÃĐtiquette extÃĐrieure conforme aux exigences du prÃĐsent rÃĻglement;
- b) les renseignements ci-aprÃĻs figurent sur lâÃĐtiquette intÃĐrieure :
- (i) le nom propre de la drogue ou, Ã dÃĐfaut, son nom usuel ou, sâil sâagit dâune drogue contenant plus dâun ingrÃĐdient actif, sa marque nominative,
- (ii) le nom du titulaire de lâidentification numÃĐrique attribuÃĐe à la drogue,
- (iii) lâactivitÃĐ de la drogue, sâil y a lieu, sauf si la drogue contient plus dâun ingrÃĐdient actif et sa marque nominative est unique en ce qui a trait à une activitÃĐ particuliÃĻre de la drogue,
- (iv) le numÃĐro de lot de la drogue,
- (v) sous rÃĐserve du paragraphe (7), la date limite dâutilisation de la drogue,
- (vi) la voie dâadministration de la drogue,
- (vii) tout renseignement nÃĐcessaire en vue de prÃĐvenir un prÃĐjudice à la santÃĐ humaine, sâil y a lieu,
- (viii) la quantitÃĐ nette de la drogue dans le rÃĐcipient.
(7) LâÃĐtiquette dâune drogue stockÃĐe en vue de son utilisation en cas dâurgence peut ne pas indiquer la date limite dâutilisation visÃĐe à lâalinÃĐa (2)e) et au sous-alinÃĐa (6)b)(v) si un autre moyen pour communiquer cette date aux individus qui administrent la drogue est fourni.
Ãtiquetage des drogues sur ordonnance
C.04.010 (1) Lâemballage dâune drogue sous forme posologique qui est une drogue sur ordonnance doit porter le symbole ÂŦ Pr Âŧ dans le quart supÃĐrieur gauche de lâespace principal des ÃĐtiquettes intÃĐrieure et extÃĐrieure ou, dans le cas dâun rÃĐcipient à dose unique, dans le quart supÃĐrieur gauche de lâespace principal de lâÃĐtiquette extÃĐrieure.
(2) Le paragraphe (1) ne sâapplique pas :
- a) aux drogues vendues au titulaire dâune licence dâÃĐtablissement dÃĐlivrÃĐe aux termes du titre 1A;
- b) aux drogues vendues sur ordonnance.
16 (1) La dÃĐfinition de drogue dÃĐsignÃĐe contre la COVID-19, à lâarticle C.08.001.1 du mÊme rÃĻglement, est abrogÃĐe.
(2) Lâarticle C.08.001.1 du mÊme rÃĻglement est modifiÃĐ par adjonction, selon lâordre alphabÃĐtique, de ce qui suit :
- drogue dâurgence de santÃĐ publique
- Drogue nouvelle dont les fins et le mode dâemploi recommandÃĐs par le fabricant ont trait à la COVID-19 ou à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique. (public health emergency drug)
- Liste dâaffections qui menacent la santÃĐ publique
- La Liste dâaffections qui menacent la santÃĐ publique au Canada, publiÃĐe sur le site Web du gouvernement du Canada, avec ses modifications successives. (List of Conditions that Threaten Public Health)
(3) La dÃĐfinition de drogue dâurgence de santÃĐ publique, à lâarticle C.08.001.1 du mÊme rÃĻglement, est remplacÃĐe par ce qui suit :
- drogue dâurgence de santÃĐ publique
- Drogue nouvelle dont les fins et le mode dâemploi recommandÃĐs par le fabricant ont trait à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique. (public health emergency drug)
17 Le mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâarticle C.08.001.1, de ce qui suit :
C.08.001.2 Le ministre ne peut ajouter une affection à la Liste dâaffections qui menacent la santÃĐ publique que sâil a des motifs raisonnables de croire, à la fois :
- a) que lâaffection prÃĐsente un risque apprÃĐciable pour la santÃĐ publique au Canada ou en rÃĐsulte;
- b) quâune intervention immÃĐdiate est nÃĐcessaire pour parer au risque.
C.08.001.3 En cas de retrait dâune affection de la Liste dâaffections qui menacent la santÃĐ publique aprÃĻs le dÃĐpÃīt, conformÃĐment aux paragraphes C.08.002(2) à (2.5) et à lâarticle C.08.005.1, dâune prÃĐsentation de drogue nouvelle à lâÃĐgard dâune drogue dâurgence de santÃĐ publique relative à cette affection, mais avant que le ministre rende une dÃĐcision dÃĐfinitive à lâÃĐgard de la prÃĐsentation en application de lâarticle C.08.004, la drogue demeure une drogue dâurgence de santÃĐ publique pour lâapplication du prÃĐsent titre tant que le ministre nâa pas rendu sa dÃĐcision.
18 (1) LâalinÃĐa C.08.002(2)o) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
- o) dans le cas dâune drogue nouvelle pour usage humain autre quâune drogue dâurgence de santÃĐ publique, une apprÃĐciation de la question de savoir si la drogue nouvelle est susceptible dâÊtre confondue avec une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe en raison de la ressemblance de la marque nominative dont lâutilisation est proposÃĐe pour cette drogue nouvelle avec la marque nominative, le nom usuel ou le nom propre de lâautre drogue.
(2) Le paragraphe C.08.002(2) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa o), de ce qui suit :
- p) dans le cas dâune drogue nouvelle pour usage humain, un plan de gestion des risques conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue, si lâune des conditions suivantes est remplie :
- (i) le degrÃĐ dâincertitude quant aux risques associÃĐs à la drogue est considÃĐrable,
- (ii) la drogue prÃĐsente un risque grave de prÃĐjudice à la santÃĐ humaine qui justifie la prise de mesures — autres que lâÃĐtiquetage — visant à diminuer la probabilitÃĐ quâun tel prÃĐjudice survienne ou à en amoindrir la gravitÃĐ.
(3) Les paragraphes C.08.002(2.1) à (2.5) du mÊme rÃĻglement sont remplacÃĐs par ce qui suit :
(2.01) Les donnÃĐes dâessais cliniques fournies dans une prÃĐsentation de drogue nouvelle au titre des alinÃĐas (2)g) ou h) ou (2.1)b) qui sont ventilÃĐes par sous-groupes de population dans une demande dâautorisation de vente de la drogue prÃĐsentÃĐe à lâAgence europÃĐenne des mÃĐdicaments ou au SecrÃĐtariat amÃĐricain aux produits alimentaires et pharmaceutiques doivent Être ventilÃĐes de la mÊme façon dans la prÃĐsentation de drogue nouvelle.
(2.1) Le fabricant peut dÃĐposer, à lâÃĐgard dâune drogue dâurgence de santÃĐ publique, une prÃĐsentation de drogue nouvelle qui nâest pas conforme aux exigences des alinÃĐas (2)g) et h) si la prÃĐsentation contient à la fois :
- a) une mention portant que la prÃĐsentation contient les preuves visÃĐes à lâalinÃĐa b);
- b) des preuves suffisantes pour conclure que les avantages associÃĐs à la drogue lâemportent sur les risques associÃĐs à cette derniÃĻre en ce qui a trait aux fins et selon le mode dâemploi recommandÃĐs, compte tenu des incertitudes dont ces avantages et ces risques font lâobjet et du besoin en matiÃĻre de santÃĐ publique relatif à la COVID-19 ou à lâaffection en cause dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique, selon le cas.
(2.2) Le fabricant peut dÃĐposer, à lâÃĐgard dâune drogue dâurgence de santÃĐ publique pour usage humain, une prÃĐsentation de drogue nouvelle qui nâest pas conforme aux exigences de lâalinÃĐa (2)j.1) si la prÃĐsentation contient une maquette de toute ÃĐtiquette à utiliser relativement à la drogue, y compris toute notice dâaccompagnement et toute documentation supplÃĐmentaire sur lâemploi de la drogue qui est fournie sur demande.
(2.3) Si, au moment de dÃĐposer sa prÃĐsentation de drogue nouvelle à lâÃĐgard dâune drogue dâurgence de santÃĐ publique, le fabricant ne peut fournir au ministre les renseignements ou le matÃĐriel visÃĐs à lâun des alinÃĐas (2)e) à k), m) et n), à lâalinÃĐa (2.1)b), au paragraphe (2.2) ou à lâarticle C.08.005.1 ou quâil fournit ces renseignements ou ce matÃĐriel mais de façon incomplÃĻte, il fournit au ministre, au mÊme moment, un plan prÃĐcisant les modalitÃĐs selon lesquelles il lui fournira les renseignements ou le matÃĐriel manquants.
(2.4) Les paragraphes (2.1) Ã (2.3) sâappliquent seulement si, Ã la fois :
- a) la prÃĐsentation de drogue nouvelle contient une mention portant que la prÃĐsentation vise une drogue dâurgence de santÃĐ publique;
- b) les fins et le mode dâemploi mentionnÃĐs dans la prÃĐsentation de drogue nouvelle ont trait uniquement à la COVID-19 ou à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique et la prÃĐsentation contient une mention à cet ÃĐgard.
(2.5) Les paragraphes (2.1) à (2.3) ne sâappliquent pas lorsque le fabricant demande un avis de conformitÃĐ Ã lâÃĐgard de la drogue dâurgence de santÃĐ publique sur la base dâune comparaison directe ou indirecte entre celle-ci et une autre drogue dâurgence de santÃĐ publique.
(4) LâalinÃĐa C.08.002(2.1)b) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
- b) des preuves suffisantes pour conclure que les avantages associÃĐs à la drogue lâemportent sur les risques associÃĐs à cette derniÃĻre en ce qui a trait aux fins et selon le mode dâemploi recommandÃĐs, compte tenu des incertitudes dont ces avantages et ces risques font lâobjet et du besoin en matiÃĻre de santÃĐ publique relatif à lâaffection applicable dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique.
(5) Le paragraphe C.08.002(2.3) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
(2.3) Si, au moment de dÃĐposer sa prÃĐsentation de drogue nouvelle à lâÃĐgard dâune drogue dâurgence de santÃĐ publique, le fabricant ne peut fournir au ministre les renseignements ou le matÃĐriel visÃĐs à lâun des alinÃĐas (2)e) à k), m), n) et p), à lâalinÃĐa (2.1)b), au paragraphe (2.2) ou à lâarticle C.08.005.1 ou quâil fournit ces renseignements ou ce matÃĐriel mais de façon incomplÃĻte, il fournit au ministre, au mÊme moment, un plan prÃĐcisant les modalitÃĐs selon lesquelles il lui fournira les renseignements ou le matÃĐriel manquants.
(6) LâalinÃĐa C.08.002(2.4)b) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
- b) les fins et le mode dâemploi mentionnÃĐs dans la prÃĐsentation de drogue nouvelle ont trait uniquement à une affection dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique et la prÃĐsentation contient une mention à cet ÃĐgard.
(7) Lâarticle C.08.002 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (3), de ce qui suit :
(4) Le fabricant peut dÃĐposer une prÃĐsentation de drogue nouvelle qui ne contient pas encore lâensemble des renseignements visÃĐs aux alinÃĐas (2)g) à i), m), n) et p) — et lâensemble des renseignements connexes à y inclure en application de lâarticle C.08.005.1, sâil y a lieu —, si les conditions suivantes sont rÃĐunies :
- a) il fournit ce qui suit au ministre avant de dÃĐposer la prÃĐsentation :
- (i) des renseignements ÃĐtablissant que les conditions prÃĐvues à lâun des alinÃĐas (5)a) à c) sont remplies,
- (ii) un plan prÃĐcisant les modalitÃĐs selon lesquelles il propose de fournir au ministre les renseignements manquants pendant la pÃĐriode de cent cinquante-cinq jours qui commence à la date du dÃĐpÃīt de la prÃĐsentation,
- (iii) des renseignements ÃĐtablissant quâil a en sa possession une quantitÃĐ apprÃĐciable de preuves en vue dâÃĐtablir lâinnocuitÃĐ de la drogue nouvelle aux fins et selon le mode dâemploi recommandÃĐs,
- (iv) des renseignements ÃĐtablissant quâil a en sa possession une quantitÃĐ apprÃĐciable de preuves en vue dâÃĐtablir lâefficacitÃĐ clinique de la drogue nouvelle aux fins et selon le mode dâemploi recommandÃĐs,
- (v) dans le cas oÃđ il demandera la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue nouvelle et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, cette identification numÃĐrique;
- b) le ministre lui fournit un avis qui prÃĐcise les modalitÃĐs selon lesquelles il doit fournir les renseignements manquants pendant la pÃĐriode de cent cinquante-cinq jours prÃĐvue au sous-alinÃĐa a)(ii);
- c) la prÃĐsentation est conforme aux exigences suivantes :
- (i) elle contient les preuves visÃĐes aux sous-alinÃĐas a)(iii) et (iv),
- (ii) son dÃĐpÃīt a lieu dans les soixante jours suivant le jour oÃđ le ministre fournit cet avis au fabricant.
(5) Pour lâapplication des sous-alinÃĐas (4)a)(i) et C.08.003(5)a)(i), les conditions sont les suivantes :
- a) la drogue nouvelle est nÃĐcessaire pour diagnostiquer, traiter, attÃĐnuer ou prÃĐvenir une maladie infectieuse ÃĐmergente qui prÃĐsente ou peut prÃĐsenter un risque grave de prÃĐjudice à la santÃĐ humaine ou animale;
- b) la drogue nouvelle est destinÃĐe à Être utilisÃĐe pour diagnostiquer, traiter, attÃĐnuer ou prÃĐvenir une maladie, un dÃĐsordre ou un ÃĐtat physique anormal qui prÃĐsente ou peut prÃĐsenter un risque grave de prÃĐjudice à la santÃĐ humaine ou animale, et les fins et le mode dâemploi recommandÃĐs de la drogue ne figurent pas parmi ceux de toute autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe;
- c) la drogue nouvelle est destinÃĐe à Être utilisÃĐe pour diagnostiquer, traiter, attÃĐnuer ou prÃĐvenir une maladie, un dÃĐsordre ou un ÃĐtat physique anormal qui prÃĐsente ou peut prÃĐsenter un risque grave de prÃĐjudice à la santÃĐ humaine ou animale et les fins et le mode dâemploi recommandÃĐs de la drogue figurent parmi ceux dâune ou de plusieurs autres drogues auxquelles une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe et nâa pas ÃĐtÃĐ annulÃĐe, mais il y a des motifs raisonnables de croire quâelle est sensiblement plus efficace ou quâelle prÃĐsente un risque sensiblement plus faible que chacune de ces autres drogues.
(6) Le ministre fournit lâavis prÃĐvu à lâalinÃĐa (4)b) au fabricant si, à la fois :
- a) le fabricant lui a fourni les renseignements visÃĐs à lâalinÃĐa (4)a);
- b) le ministre nâa pas de motifs de croire que le fabricant ne sera pas en mesure de lui fournir les renseignements manquants conformÃĐment à lâavis.
(7) La prÃĐsentation de drogue nouvelle dÃĐposÃĐe conformÃĐment au paragraphe (4) est considÃĐrÃĐe comme ayant ÃĐtÃĐ annulÃĐe par le fabricant dans les cas suivants :
- a) il nâa pas fourni au ministre certains des renseignements mentionnÃĐs dans lâavis prÃĐvu à lâalinÃĐa (4)b) dans les dix jours suivant celle des dates prÃĐcisÃĐes dans lâavis qui sâapplique, ou dans tout dÃĐlai plus long prÃĐcisÃĐ par le ministre, ou il ne sera pas en mesure de le faire;
- b) il demande la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue nouvelle et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, mais il a omis de fournir cette identification numÃĐrique au ministre avant de dÃĐposer la prÃĐsentation.
(8) Le fabricant dâune drogue nouvelle pour usage vÃĐtÃĐrinaire seulement peut dÃĐposer, à lâÃĐgard de celle-ci, une prÃĐsentation de drogue nouvelle qui ne contient pas encore lâensemble des renseignements visÃĐs aux alinÃĐas (2)d) à j), aux sous-alinÃĐas (2)k)(ii) et (iv) et aux alinÃĐas (2)m) et n) — et lâensemble des renseignements connexes à y inclure en application de lâarticle C.08.005.1, sâil y a lieu —, si les conditions suivantes sont rÃĐunies :
- a) le ministre lui a indiquÃĐ son intention dâexaminer la prÃĐsentation avec une autoritÃĐ rÃĐglementaire ÃĐtrangÃĻre au sens du paragraphe C.10.001(1);
- b) au moment du dÃĐpÃīt, la prÃĐsentation contient ce qui suit :
- (i) un plan prÃĐcisant les modalitÃĐs selon lesquelles il fournira au ministre les renseignements manquants,
- (ii) à tout le moins certains des renseignements visÃĐs à lâun des alinÃĐas (2)d) à h), m) et n),
- (iii) dans le cas oÃđ le fabricant demande la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue nouvelle et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, cette identification numÃĐrique.
(9) La prÃĐsentation de drogue nouvelle dÃĐposÃĐe conformÃĐment au paragraphe (8) est considÃĐrÃĐe comme ayant ÃĐtÃĐ annulÃĐe par le fabricant dans les cas suivants :
- a) il avise le ministre quâil ne veut plus que ce dernier examine la prÃĐsentation avec lâautoritÃĐ rÃĐglementaire ÃĐtrangÃĻre;
- b) il demande la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue en question et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, mais il a omis de fournir cette identification numÃĐrique au ministre au moment de dÃĐposer la prÃĐsentation.
19 LâalinÃĐa C.08.002.01(2)b) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le sous-alinÃĐa (x), de ce qui suit :
- (xi) un plan de gestion des risques conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue nouvelle.
20 Le paragraphe C.08.002.1(2) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa a), de ce qui suit :
- a.1) dans le cas dâune drogue nouvelle pour usage humain, un plan de gestion des risques conforme aux exigences de lâarticle C.01.700 à lâÃĐgard de la drogue, si lâune des conditions prÃĐvues aux sous-alinÃĐas C.08.002(2)p)(i) et (ii) est remplie;
21 (1) Le paragraphe C.08.003(2) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa b), de ce qui suit :
- b.1) dans le cas dâun vaccin contre lâinfluenza mentionnÃĐ dans la Liste des vaccins contre lâinfluenza pouvant faire lâobjet de supplÃĐments à une prÃĐsentation de drogue nouvelle, publiÃĐe sur le site Web du gouvernement du Canada, avec ses modifications successives, les souches auxquelles le vaccin se rapporte;
(2) Lâarticle C.08.003 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (3), de ce qui suit :
(3.01) Les donnÃĐes dâessais cliniques fournies dans le supplÃĐment à une prÃĐsentation de drogue nouvelle qui sont ventilÃĐes par sous-groupes de population dans une demande dâautorisation de vente de la drogue prÃĐsentÃĐe à lâAgence europÃĐenne des mÃĐdicaments ou au SecrÃĐtariat amÃĐricain aux produits alimentaires et pharmaceutiques doivent Être ventilÃĐes de la mÊme façon dans le supplÃĐment.
(3) Lâarticle C.08.003 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs le paragraphe (4), de ce qui suit :
(5) Le fabricant dâune drogue nouvelle à lâÃĐgard de laquelle une prÃĐsentation de drogue nouvelle a ÃĐtÃĐ dÃĐposÃĐe peut dÃĐposer un supplÃĐment à cette prÃĐsentation qui ne contient pas encore lâensemble des renseignements et du matÃĐriel visÃĐs au paragraphe (3) — et lâensemble des renseignements connexes à y inclure en application de lâarticle C.08.005.1, sâil y a lieu —, si les conditions suivantes sont rÃĐunies :
- a) il fournit au ministre ce qui suit avant de dÃĐposer le supplÃĐment :
- (i) des renseignements ÃĐtablissant que les conditions prÃĐvues à lâun des alinÃĐas C.08.002(5)a) à c) sont remplies,
- (ii) un plan prÃĐcisant les modalitÃĐs selon lesquelles il propose de fournir au ministre les renseignements ou le matÃĐriel manquants pendant la pÃĐriode de cent cinquante-cinq jours qui commence à la date du dÃĐpÃīt du supplÃĐment,
- (iii) des renseignements ÃĐtablissant quâil a en sa possession une quantitÃĐ apprÃĐciable des renseignements et du matÃĐriel visÃĐs au paragraphe (3),
- (iv) dans le cas oÃđ il demandera la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue en question et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, cette identification numÃĐrique;
- b) le ministre lui fournit un avis qui prÃĐcise les modalitÃĐs selon lesquelles il doit fournir les renseignements ou le matÃĐriel manquants pendant la pÃĐriode de cent cinquante-cinq jours prÃĐvue au sous-alinÃĐa a)(ii);
- c) le supplÃĐment est conforme aux exigences suivantes :
- (i) il contient les renseignements et le matÃĐriel visÃĐs au paragraphe (3) que le fabricant avait en sa possession au moment de fournir des renseignements au ministre au titre du sous-alinÃĐa a)(iii),
- (ii) son dÃĐpÃīt a lieu dans les soixante jours suivant le jour oÃđ le ministre fournit cet avis au fabricant.
(6) Le ministre fournit lâavis prÃĐvu à lâalinÃĐa (5)b) au fabricant si, à la fois :
- a) le fabricant lui a fourni les renseignements visÃĐs à lâalinÃĐa (5)a);
- b) le ministre nâa pas de motifs de croire que le fabricant ne sera pas en mesure de lui fournir les renseignements ou le matÃĐriel manquants conformÃĐment à lâavis.
(7) Le supplÃĐment dÃĐposÃĐ conformÃĐment au paragraphe (5) est considÃĐrÃĐ comme ayant ÃĐtÃĐ annulÃĐ par le fabricant dans les cas suivants :
- a) il nâa pas fourni au ministre certains des renseignements ou du matÃĐriel mentionnÃĐs dans lâavis prÃĐvu à lâalinÃĐa (5)b) dans les dix jours suivant celle des dates prÃĐcisÃĐes dans lâavis qui sâapplique, ou dans tout dÃĐlai plus long prÃĐcisÃĐ par le ministre, ou il ne sera pas en mesure de le faire;
- b) il demande la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue nouvelle et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, mais il a omis de fournir cette identification numÃĐrique au ministre avant de dÃĐposer le supplÃĐment.
(8) Le fabricant dâune drogue nouvelle pour usage vÃĐtÃĐrinaire seulement à lâÃĐgard de laquelle une prÃĐsentation de drogue nouvelle a ÃĐtÃĐ dÃĐposÃĐe peut dÃĐposer un supplÃĐment à cette prÃĐsentation qui ne contient pas encore lâensemble des renseignements et du matÃĐriel visÃĐs au paragraphe (3) — et lâensemble des renseignements connexes à y inclure en application de lâarticle C.08.005.1, sâil y a lieu —, si les conditions suivantes sont rÃĐunies :
- a) le ministre lui a indiquÃĐ son intention dâexaminer le supplÃĐment avec une autoritÃĐ rÃĐglementaire ÃĐtrangÃĻre au sens du paragraphe C.10.001(1);
- b) au moment du dÃĐpÃīt, le supplÃĐment contient ce qui suit :
- (i) un plan prÃĐcisant les modalitÃĐs selon lesquelles il fournira au ministre les renseignements ou le matÃĐriel manquants,
- (ii) certains des renseignements ou du matÃĐriel visÃĐs au paragraphe (3),
- (iii) dans le cas oÃđ il demande la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue en question et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, cette identification numÃĐrique.
(9) Le supplÃĐment dÃĐposÃĐ conformÃĐment au paragraphe (8) est considÃĐrÃĐ comme ayant ÃĐtÃĐ annulÃĐ par le fabricant dans les cas suivants :
- a) il avise le ministre quâil ne veut plus que ce dernier examine le supplÃĐment avec lâautoritÃĐ rÃĐglementaire ÃĐtrangÃĻre;
- b) il demande la dÃĐlivrance de lâavis de conformitÃĐ sur la base dâune comparaison directe ou indirecte entre la drogue en question et une autre drogue à laquelle une identification numÃĐrique a ÃĐtÃĐ attribuÃĐe, mais il a omis de fournir cette identification numÃĐrique au ministre au moment de dÃĐposer le supplÃĐment.
(10) Le fabricant dâun vaccin mentionnÃĐ dans la liste mentionnÃĐe à lâalinÃĐa (2)b.1) peut dÃĐposer un supplÃĐment à la prÃĐsentation de drogue nouvelle dÃĐposÃĐe à lâÃĐgard du vaccin qui ne contient pas encore lâensemble des renseignements et du matÃĐriel visÃĐs au paragraphe (3) — et lâensemble des renseignements connexes à y inclure en application de lâarticle C.08.005.1, sâil y a lieu —, si le supplÃĐment se rapporte à lâÃĐlÃĐment visÃĐ Ã cet alinÃĐa.
22 Lâarticle C.08.003.1 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
C.08.003.1 Dans le cadre de son examen dâune prÃĐsentation de drogue nouvelle, dâune prÃĐsentation de drogue nouvelle pour usage exceptionnel, dâune prÃĐsentation abrÃĐgÃĐe de drogue nouvelle, dâune prÃĐsentation abrÃĐgÃĐe de drogue nouvelle pour usage exceptionnel ou dâun supplÃĐment à lâune de ces prÃĐsentations, le ministre peut examiner les renseignements ou le matÃĐriel ci-aprÃĻs pour ÃĐvaluer lâinnocuitÃĐ et lâefficacitÃĐ de la drogue nouvelle visÃĐe par la prÃĐsentation ou le supplÃĐment :
- a) ceux que toute personne a fournis sous le rÃĐgime de la Loi;
- b) ceux obtenus de tout lieu de fabrication, dâemballage, dâÃĐtiquetage ou dâanalyse — en cours ou à venir — de la drogue nouvelle ou de tout ingrÃĐdient actif de celle-ci au sens du paragraphe C.01A.001(1);
- c) ceux obtenus — directement ou indirectement — dâune autoritÃĐ rÃĐglementaire ÃĐtrangÃĻre au sens du paragraphe C.10.001(1).
23 Le mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâarticle C.08.003.1, de ce qui suit :
C.08.003.2 Il est entendu que le fabricant peut Être assujetti à lâobligation de fournir le plan de gestion des risques visÃĐ Ã lâalinÃĐa C.08.002(2)p), au sous-alinÃĐa C.08.002.01(2)b)(ix) ou à lâalinÃĐa C.08.002.1(2)a.1) à tout moment avant que le ministre rende une dÃĐcision dÃĐfinitive en application des articles C.08.004 ou C.08.004.01, selon le cas.
24 Lâintertitre prÃĐcÃĐdant lâarticle C.08.009.01 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
PrÃĐpositionnement de drogues dâurgence de santÃĐ publique
25 Lâarticle C.08.009.01 du mÊme rÃĻglement est modifiÃĐ par adjonction, selon lâordre alphabÃĐtique, de ce qui suit :
- classe de forme posologique
- Sâentend au sens du paragraphe C.01A.001(1). (dosage form class)
- emballer-ÃĐtiqueter
- Sâentend au sens du paragraphe C.01A.001(1). (package/label)
- manufacturer
- Sâentend au sens du paragraphe C.01A.001(1). (fabricate)
26 Lâarticle C.08.009.02 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
C.08.009.02 Les articles C.08.009.03 à C.08.009.05 sâappliquent à lâÃĐgard dâune drogue dâurgence de santÃĐ publique si les conditions suivantes sont rÃĐunies :
- a) aucun avis de conformitÃĐ nâa ÃĐtÃĐ dÃĐlivrÃĐ Ã son ÃĐgard en application des articles C.08.004 ou C.08.004.01;
- b) Sa MajestÃĐ du chef du Canada a conclu un contrat en vue de son acquisition.
27 Le paragraphe C.08.009.03(1) du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
C.08.009.03 (1) Le titulaire dâune licence dâÃĐtablissement peut importer une drogue dâurgence de santÃĐ publique si les conditions suivantes sont rÃĐunies :
- a) lâadministrateur en chef de la santÃĐ publique fournit au ministre :
- (i) de lâinformation selon laquelle la drogue fait lâobjet dâune prÃĐsentation de drogue nouvelle ou dâune demande dâautorisation de vente prÃĐsentÃĐe à une autoritÃĐ rÃĐglementaire ÃĐtrangÃĻre,
- (ii) le nom et la description de la drogue,
- (iii) les nom et coordonnÃĐes du fabricant de la drogue,
- (iv) de lâinformation quant à la quantitÃĐ de la drogue à importer,
- (v) les nom et coordonnÃĐes du titulaire de licence,
- (vi) lâadresse municipale du lieu oÃđ la drogue sera entreposÃĐe aprÃĻs lâimportation;
- b) le titulaire de licence fournit au ministre :
- (i) les nom et coordonnÃĐes de chaque manufacturier, emballeur-ÃĐtiqueteur et analyste de la drogue et lâadresse municipale de chaque bÃĒtiment oÃđ celle-ci sera manufacturÃĐe, emballÃĐe-ÃĐtiquetÃĐe ou analysÃĐe, avec indication, pour chaque bÃĒtiment, de ce qui suit :
- (A) les activitÃĐs mentionnÃĐes au tableau I de lâarticle C.01A.008 qui sâappliquent à la drogue,
- (B) les catÃĐgories mentionnÃĐes au tableau II de cet article qui sâappliquent à la drogue,
- (C) pour chacune de ces catÃĐgories, la classe de forme posologique, le cas ÃĐchÃĐant, et une mention indiquant sâil sâagit dâune drogue stÃĐrile,
- (ii) le certificat dâun inspecteur indiquant que les bÃĒtiments, lâÃĐquipement et les mÃĐthodes et pratiques de chaque manufacturier, emballeur-ÃĐtiqueteur et analyste satisfont aux exigences applicables des titres 2 à 4 ou, à dÃĐfaut, toute autre preuve ÃĐtablissant quâil est satisfait à ces exigences;
- (i) les nom et coordonnÃĐes de chaque manufacturier, emballeur-ÃĐtiqueteur et analyste de la drogue et lâadresse municipale de chaque bÃĒtiment oÃđ celle-ci sera manufacturÃĐe, emballÃĐe-ÃĐtiquetÃĐe ou analysÃĐe, avec indication, pour chaque bÃĒtiment, de ce qui suit :
- c) la drogue appartient à une catÃĐgorie de drogues que la licence permet dâimporter.
28 (1) Le passage de lâarticle C.08.009.04 du mÊme rÃĻglement prÃĐcÃĐdant lâalinÃĐa a) est remplacÃĐ par ce qui suit :
C.08.009.04 Les articles A.01.040, C.01.004.1 et C.01A.006 et, à lâexception des dispositions ci-aprÃĻs, les titres 2 à 4 ne sâappliquent pas à lâÃĐgard de lâimportation, au titre de lâarticle C.08.009.03, dâune drogue dâurgence de santÃĐ publique :
(2) Lâarticle C.08.009.04 du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa b), de ce qui suit :
- b.1) lâarticle C.02.012.1 en ce qui a trait à lâentreposage de la drogue par le titulaire de licence;
(3) LâalinÃĐa C.08.009.04j) du mÊme rÃĻglement est abrogÃĐ.
29 Le passage de lâarticle C.08.009.05 du mÊme rÃĻglement prÃĐcÃĐdant lâalinÃĐa a) est remplacÃĐ par ce qui suit :
C.08.009.05 MalgrÃĐ toute disposition du prÃĐsent rÃĻglement, le titulaire dâune licence dâÃĐtablissement peut distribuer une drogue dâurgence de santÃĐ publique quâil a importÃĐe en vertu de lâarticle C.08.009.03 si les conditions suivantes sont rÃĐunies :
30 (1) Le paragraphe C.08.011.2(2) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa c), de ce qui suit :
- c.1) lâarticle C.02.012.1 en ce qui a trait à lâentreposage de la drogue nouvelle par le titulaire dâune licence dâÃĐtablissement;
(2) LâalinÃĐa C.08.011.2(2)k) du mÊme rÃĻglement est abrogÃĐ.
31 (1) Le paragraphe C.10.001(5) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa c), de ce qui suit :
- c.1) lâarticle C.02.012.1 en ce qui a trait à lâentreposage de la drogue par le titulaire;
(2) LâalinÃĐa C.10.001(5)k) du mÊme rÃĻglement est abrogÃĐ.
32 Le paragraphe C.10.002(2) du mÊme rÃĻglement est modifiÃĐ par adjonction, aprÃĻs lâalinÃĐa c), de ce qui suit :
- c.1) lâarticle C.02.012.1 en ce qui a trait à lâentreposage de la drogue par le titulaire;
33 Dans les passages ci-aprÃĻs du mÊme rÃĻglement, ÂŦ drogue dÃĐsignÃĐe contre la COVID-19 Âŧ est remplacÃĐ par ÂŦ drogue Âŧ :
- a) les alinÃĐas C.08.009.04a), d) et e);
- b) les alinÃĐas C.08.009.05a) et b).
RÃĻglement sur les instruments mÃĐdicaux
34 Les paragraphes 36(2) Ã (4) du RÃĻglement sur les instruments mÃĐdicaux rÃĐfÃĐrence 35 sont remplacÃĐs par ce qui suit :
(2) Le ministre peut, en tout temps, assortir de conditions lâhomologation dÃĐlivrÃĐe à lâÃĐgard dâun instrument mÃĐdical ou modifier de telles conditions aprÃĻs avoir pris en compte les facteurs suivants :
- a) la question de savoir si les avantages ou les risques associÃĐs à lâinstrument font lâobjet dâincertitudes;
- b) la question de savoir si les exigences prÃĐvues sous le rÃĐgime de la Loi sont suffisantes pour atteindre les objectifs suivants :
- (i) maintenir la sÃŧretÃĐ et lâefficacitÃĐ de lâinstrument,
- (ii) optimiser les avantages et gÃĐrer les risques associÃĐs à lâinstrument,
- (iii) dÃĐceler tout changement et gÃĐrer les incertitudes dont les avantages et les risques associÃĐs à lâinstrument font lâobjet;
- c) la question de savoir si les conditions projetÃĐes peuvent contribuer à lâatteinte des objectifs mentionnÃĐs aux sous-alinÃĐas b)(i) à (iii);
- d) la question de savoir si le respect des conditions projetÃĐes est rÃĐalisable sur le plan technique;
- e) la question de savoir sâil existe des moyens moins exigeants pour atteindre les objectifs des conditions projetÃĐes.
35 Lâarticle 37 du mÊme rÃĻglement est remplacÃĐ par ce qui suit :
37 Si les conditions dont lâhomologation dâun instrument diagnostique in vitro est assortie prÃĐvoient que des essais soient effectuÃĐs pour veiller à ce que lâinstrument satisfasse toujours aux exigences applicables prÃĐvues aux articles 10 à 20, il est interdit de vendre tout instrument provenant de tout lot de lâinstrument diagnostique in vitro sauf si, à la fois :
- a) le protocole dâessai et les rÃĐsultats des essais effectuÃĐs sur des instruments faisant partie du lot ont ÃĐtÃĐ fournis au ministre;
- b) le ministre conclut, selon les renseignements qui lui ont ÃĐtÃĐ fournis en application de lâalinÃĐa a), que les instruments faisant partie du lot satisfont toujours aux exigences applicables prÃĐvues aux articles 10 Ã 20.
RÃĻglement modifiant le RÃĻglement sur les aliments et drogues (ArrÊtÃĐ dâurgence concernant lâimportation, la vente et la publicitÃĐ de drogues à utiliser relativement à la COVID-19)
36 LâalinÃĐa 20e) du RÃĻglement modifiant le RÃĻglement sur les aliments et drogues (ArrÊtÃĐ dâurgence concernant lâimportation, la vente et la publicitÃĐ de drogues à utiliser relativement à la COVID-19) rÃĐfÃĐrence 36 est remplacÃĐ par ce qui suit :
- e) les articles C.04.004, C.04.005, C.04.007, C.04.009 et C.04.010.
Dispositions transitoires
37 Sauf indication contraire du contexte, les termes utilisÃĐs dans les articles 38 Ã 43 sâentendent au sens du RÃĻglement sur les aliments et drogues.
38 (1) Les conditions dont toute identification numÃĐrique est assortie en vertu du paragraphe C.01.014.21(1.1) du RÃĻglement sur les aliments et drogues, dans sa version antÃĐrieure à la date dâentrÃĐe en vigueur du paragraphe 6(1) du prÃĐsent rÃĻglement, qui sâappliquent immÃĐdiatement avant cette date continuent de sâappliquer et peuvent Être modifiÃĐes par le ministre en vertu du paragraphe C.01.014.21(1.1) du RÃĻglement sur les aliments et drogues, dans sa version à cette date ou aprÃĻs celle-ci.
(2) Les conditions dont toute identification numÃĐrique est assortie en vertu des paragraphes C.01.014.21(1) ou (1.1) du RÃĻglement sur les aliments et drogues, dans leur version antÃĐrieure à la date dâentrÃĐe en vigueur du paragraphe 6(4) du prÃĐsent rÃĻglement, qui sâappliquent immÃĐdiatement avant cette date continuent de sâappliquer et peuvent Être modifiÃĐes par le ministre en vertu de lâarticle C.01.014.21 du RÃĻglement sur les aliments et drogues, dans sa version à cette date ou aprÃĻs celle-ci.
39 (1) Les mentions de ÂŦ plan existant Âŧ à lâarticle C.01.702 et au paragraphe C.01.703(1) du RÃĻglement sur les aliments et drogues valent mention, dans le cas dâune drogue à lâÃĐgard de laquelle un plan de gestion des risques a ÃĐtÃĐ fourni au ministre avant la date dâentrÃĐe en vigueur de lâarticle 7 du prÃĐsent rÃĻglement, de la version la plus rÃĐcente du plan.
(2) Le paragraphe (1) cesse de sâappliquer à lâÃĐgard dâune drogue lorsquâun plan de gestion des risques à jour est fourni au ministre à lâÃĐgard de la drogue conformÃĐment aux articles C.01.702 ou C.01.703 du RÃĻglement sur les aliments et drogues.
40 Dans le cas oÃđ une demande de dÃĐlivrance ou de modification dâune licence dâÃĐtablissement qui contient, conformÃĐment aux paragraphes C.01A.005(2) ou C.01A.006(1.1) du RÃĻglement sur les aliments et drogues, une mention faisant rÃĐfÃĐrence à la COVID-19 est prÃĐsentÃĐe au ministre avant la date dâentrÃĐe en vigueur des paragraphes 8(2) et 9(2) du prÃĐsent rÃĻglement, le ministre peut dÃĐlivrer ou modifier la licence au titre de cette demande à cette date ou aprÃĻs celle-ci, malgrÃĐ les modifications apportÃĐes par ces paragraphes.
41 (1) Le paragraphe C.08.002(2.01) du RÃĻglement sur les aliments et drogues ne sâapplique pas à lâÃĐgard de la prÃĐsentation de drogue nouvelle qui a ÃĐtÃĐ dÃĐposÃĐe avant la date dâentrÃĐe en vigueur de ce paragraphe.
(2) Le paragraphe C.08.003(3.01) du RÃĻglement sur les aliments et drogues ne sâapplique pas à lâÃĐgard du supplÃĐment à une prÃĐsentation de drogue nouvelle qui a ÃĐtÃĐ dÃĐposÃĐ avant la date dâentrÃĐe en vigueur de ce paragraphe.
42 (1) Si, à la date dâentrÃĐe en vigueur du paragraphe 16(1) du prÃĐsent rÃĻglement, le ministre nâa pas rendu de dÃĐcision dÃĐfinitive en application de lâarticle C.08.004 du RÃĻglement sur les aliments et drogues à lâÃĐgard de la prÃĐsentation de drogue nouvelle dÃĐposÃĐe avant cette date conformÃĐment aux paragraphes C.08.002(2) à (2.5) et à lâarticle C.08.005.1 de ce rÃĻglement à lâÃĐgard dâune drogue dÃĐsignÃĐe contre la COVID-19 au sens de lâarticle C.08.001.1 du mÊme rÃĻglement, dans sa version avant cette date, les paragraphes C.08.002(2) à (2.5), dans leur version antÃĐrieure à cette date, continuent de sâappliquer à lâÃĐgard de la prÃĐsentation jusquâà ce que le ministre rende une dÃĐcision dÃĐfinitive.
(2) Si, à la date dâentrÃĐe en vigueur du paragraphe 16(3) du prÃĐsent rÃĻglement, le ministre nâa pas rendu de dÃĐcision dÃĐfinitive en application de lâarticle C.08.004 du RÃĻglement sur les aliments et drogues à lâÃĐgard de la prÃĐsentation de drogue nouvelle dÃĐposÃĐe avant cette date conformÃĐment aux paragraphes C.08.002(2) à (2.5) et à lâarticle C.08.005.1 de ce rÃĻglement à lâÃĐgard dâune drogue dâurgence de santÃĐ publique au sens de lâarticle C.08.001.1 du mÊme rÃĻglement, dans sa version avant cette date, relative à la COVID-19, la drogue demeure une drogue dâurgence de santÃĐ publique pour lâapplication du titre 8 de la partie C du mÊme rÃĻglement jusquâà ce que :
- a) le ministre rende sa dÃĐcision dÃĐfinitive;
- b) dans le cas oÃđ la COVID-19 est dÃĐcrite dans la Liste dâaffections qui menacent la santÃĐ publique au sens de lâarticle C.08.001.1 du mÊme rÃĻglement au moment oÃđ le ministre rend sa dÃĐcision dÃĐfinitive, la COVID-19 soit retirÃĐe de cette liste.
43 (1) Si, à la date dâentrÃĐe en vigueur du paragraphe 18(7) du prÃĐsent rÃĻglement ou avant celle-ci, le ministre indique au fabricant dâune drogue nouvelle pour usage vÃĐtÃĐrinaire seulement qui a dÃĐposÃĐ, à lâÃĐgard de cette drogue, une prÃĐsentation de drogue nouvelle qui ne contient pas encore lâensemble des renseignements et du matÃĐriel à y inclure son intention dâexaminer la prÃĐsentation avec une autoritÃĐ rÃĐglementaire ÃĐtrangÃĻre, le fabricant lui fournit, au plus tard soixante jours aprÃĻs cette date, un plan prÃĐcisant les modalitÃĐs selon lesquelles il lui fournira les renseignements ou le matÃĐriel manquants, à moins quâil lui ait fourni un tel plan à cette date ou avant celle-ci.
(2) Si, à la date dâentrÃĐe en vigueur du paragraphe 21(3) du prÃĐsent rÃĻglement ou avant celle-ci, le ministre indique au fabricant dâune drogue nouvelle pour usage vÃĐtÃĐrinaire seulement qui a dÃĐposÃĐ un supplÃĐment à la prÃĐsentation de drogue nouvelle dÃĐposÃĐe à lâÃĐgard de la drogue qui ne contient pas encore lâensemble des renseignements et du matÃĐriel à y inclure son intention dâexaminer le supplÃĐment avec une autoritÃĐ rÃĐglementaire ÃĐtrangÃĻre, le fabricant lui fournit un plan prÃĐcisant les modalitÃĐs selon lesquelles il lui fournira les renseignements ou le matÃĐriel manquants au plus tard soixante jours aprÃĻs cette date, à moins quâil lui ait fourni un tel plan à cette date ou avant celle-ci.
44 Les conditions dont toute homologation visÃĐe au paragraphe 36(1) du RÃĻglement sur les instruments mÃĐdicaux est assortie avant la date dâentrÃĐe en vigueur de lâarticle 34 du prÃĐsent rÃĻglement sont rÃĐputÃĐes Être imposÃĐes par le ministre en vertu du paragraphe 36(2) du RÃĻglement sur les instruments mÃĐdicaux, dans sa version à cette date.
EntrÃĐe en vigueur
45 (1) Sous rÃĐserve des paragraphes (2) et (3), le prÃĐsent rÃĻglement entre en vigueur le jour de son enregistrement.
(2) Les dispositions ci-aprÃĻs entrent en vigueur au premier anniversaire de lâenregistrement du prÃĐsent rÃĻglement :
- a) les articles 4 et 5;
- b) le paragraphe 6(4);
- c) lâarticle 7;
- d) les paragraphes 18(2), (5) et (7);
- e) les articles 19 et 20;
- f) les paragraphes 21(1) et (3);
- g) lâarticle 23;
- h) les articles 34 et 35.
(3) Les dispositions ci-aprÃĻs entrent en vigueur à la date fixÃĐe par modification du prÃĐsent paragraphe :
- a) les paragraphes 8(2), 9(2) et 10(2);
- b) le paragraphe 16(3);
- c) les paragraphes 18(4) et (6).
Conditions dâutilisation et Avis de confidentialitÃĐ
Conditions d’utilisation
Vous Êtes tenu de vous assurer que les commentaires que vous formulez ne contiennent aucun des ÃĐlÃĐments suivants :
- renseignement personnel;
- renseignement protÃĐgÃĐ ou classifiÃĐ du gouvernement du Canada;
- commentaire discriminatoire ou qui incite à la discrimination fondÃĐe sur la race, le sexe, la religion, l’orientation sexuelle ou contre tout autre groupe protÃĐgÃĐ en vertu de la Loi canadienne sur les droits de la personne ou de la Charte canadienne des droits et libertÃĐs;
- commentaire haineux, diffamatoire ou obscÃĻne;
- commentaire menaçant, violent, intimidant ou harcelant;
- commentaire venant à l’encontre des lois fÃĐdÃĐrales, provinciales ou territoriales du Canada;
- commentaire qui constitue une usurpation d’identitÃĐ, de la publicitÃĐ ou du pollupostage;
- commentaire dont le but est d’encourager ou d’inciter une activitÃĐ criminelle;
- commentaire rÃĐdigÃĐ dans une langue autre que le français ou l’anglais;
- commentaire qui contrevient autrement au prÃĐsent avis.
L’institution fÃĐdÃĐrale qui gÃĻre le changement rÃĐglementaire proposÃĐ conserve le droit d’examiner et de supprimer les renseignements personnels, les propos haineux ou tout autre renseignement jugÃĐ inappropriÃĐ Ã la publication, tel qu’il est dÃĐcrit ci-dessus.
Les renseignements commerciaux confidentiels ne doivent Être affichÃĐs que dans la zone de texte rÃĐservÃĐe à cette fin. En gÃĐnÃĐral, ÂŦ renseignements commerciaux confidentiels Âŧ dÃĐsigne les renseignements qui i) ne sont pas accessibles au public, ii) sont traitÃĐs de façon confidentielle par la personne dont l’entreprise est concernÃĐe par ces renseignements et iii) ont une valeur ÃĐconomique rÃĐelle ou potentielle pour la personne ou ses concurrents, car ils ne sont pas accessibles au public et leur divulgation entraÃŪnerait une perte financiÃĻre pour la personne ou un gain important pour ses concurrents. Les commentaires fournis dans la zone rÃĐservÃĐe aux renseignements commerciaux confidentiels qui correspondent à cette description ne seront pas rendus publics. L’institution fÃĐdÃĐrale qui gÃĻre le changement rÃĐglementaire proposÃĐ conserve le droit de rendre le commentaire public s’il n’est pas considÃĐrÃĐ qu’il s’agit d’un renseignement commercial confidentiel.
Vos commentaires seront affichÃĐs sur le site Web de la Gazette du Canada à la disposition du public pour examen. Cependant, vous avez le droit de soumettre vos commentaires de façon anonyme. Le cas ÃĐchÃĐant, vos commentaires seront rendus publics et attribuÃĐs à une personne anonyme. Aucun autre renseignement à votre sujet ne sera rendu public.
Les commentaires seront affichÃĐs sur le site Web de la Gazette du Canada pendant au moins 10 ans.
à l’heure actuelle, la fonction de commentaires en ligne ne prend pas en charge les piÃĻces jointes; les zones de texte ne prennent pas en charge les graphiques, les tableaux ou autres ÃĐlÃĐments multimÃĐdias semblables. Si vous devez joindre une piÃĻce jointe à vos commentaires, veuillez ÃĐcrire à l’adresse de courriel ministÃĐrielle indiquÃĐe dans l’avis de publication prÃĐalable. Veuillez noter que la communication par courriel public n’est pas sÃĐcurisÃĐe. Par consÃĐquent, si la piÃĻce jointe à transmettre contient des renseignements de nature dÃĐlicate, veuillez ÃĐcrire à l’adresse de courriel ministÃĐrielle pour discuter des façons dont vous pouvez transmettre ces renseignements.
Avis de confidentialitÃĐ
Les renseignements fournis sont recueillis en vertu de la Loi sur la gestion des finances publiques, de la Loi sur le ministÃĻre des Travaux publics et des Services gouvernementaux, de la Loi de mise en Åuvre de l’Accord Canada–Ãtats-Unis–Mexique, ainsi que des lois habilitantes des organismes de rÃĐglementation concernÃĐs, aux fins de recueillir des commentaires liÃĐs aux changements rÃĐglementaires. Vos commentaires et vos documents sont recueillis dans le but d’accroÃŪtre la transparence du processus rÃĐglementaire et de rendre le gouvernement plus accessible aux Canadiens.
Les renseignements personnels soumis sont recueillis, utilisÃĐs, communiquÃĐs, conservÃĐs et protÃĐgÃĐs contre l’accÃĻs par les personnes ou les organismes non autorisÃĐs conformÃĐment aux dispositions de la Loi sur la protection des renseignements personnels et du RÃĻglement sur la protection des renseignements personnels. Les noms des personnes fournis ne seront pas affichÃĐs en ligne; ils seront toutefois conservÃĐs pour que nous puissions communiquer avec ces personnes au besoin. Les noms des organisations qui formulent des commentaires seront affichÃĐs en ligne.
Les renseignements soumis, y compris les renseignements personnels, seront accessibles à Services publics et Approvisionnement Canada, à qui incombe les responsabilitÃĐs de la page Web de la Gazette du Canada, et à l’institution fÃĐdÃĐrale responsable de la gestion du changement rÃĐglementaire proposÃĐ.
Toute personne est en droit de demander que les renseignements personnels la concernant lui soient communiquÃĐs ou qu’ils soient corrigÃĐs. Pour demander l’accÃĻs à vos renseignements personnels ou leur correction, communiquez avec le Bureau de l’accÃĻs à l’information et de la protection des renseignements personnels (AIPRP) de l’institution fÃĐdÃĐrale responsable de la gestion du changement rÃĐglementaire proposÃĐ.
Vous pouvez adresser une plainte au Commissariat à la protection de la vie privÃĐe du Canada au sujet de la gestion de vos renseignements personnels par une institution fÃĐdÃĐrale.
Les renseignements personnels fournis sont versÃĐs dans le fichier de renseignements personnels POU 938 ActivitÃĐs de sensibilisation. Les personnes qui souhaitent accÃĐder à leurs renseignements personnels en vertu de la Loi sur la protection des renseignements personnels doivent en faire la demande à l’organisme de rÃĐglementation compÃĐtent en fournissant suffisamment de renseignements pour permettre à l’institution fÃĐdÃĐrale de rÃĐcupÃĐrer les renseignements personnels concernant ces personnes. L’institution fÃĐdÃĐrale pourrait avoir de la difficultÃĐ Ã retracer les renseignements personnels au sujet de personnes qui formulent des commentaires de façon anonyme et qui demandent l’accÃĻs à leurs renseignements personnels.